 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Acta Botanica Sinica, Vol. 45, No. 7, 2003, pp. 766-769 Rapid Communication Matrix Generator (MG): a Program for Creating 0/1 Matrix from Sized DNA Fragments ZHOU Shi-Liang1 *, Peter QIAN2 (1. Laboratory of Systematic and Evolutionary Botany, Institute
of Botany, The Chinese Academy of
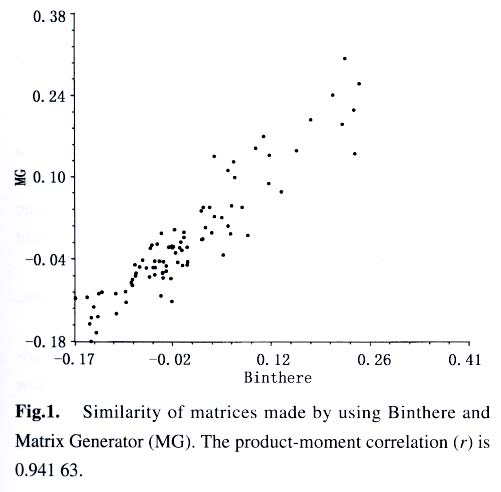
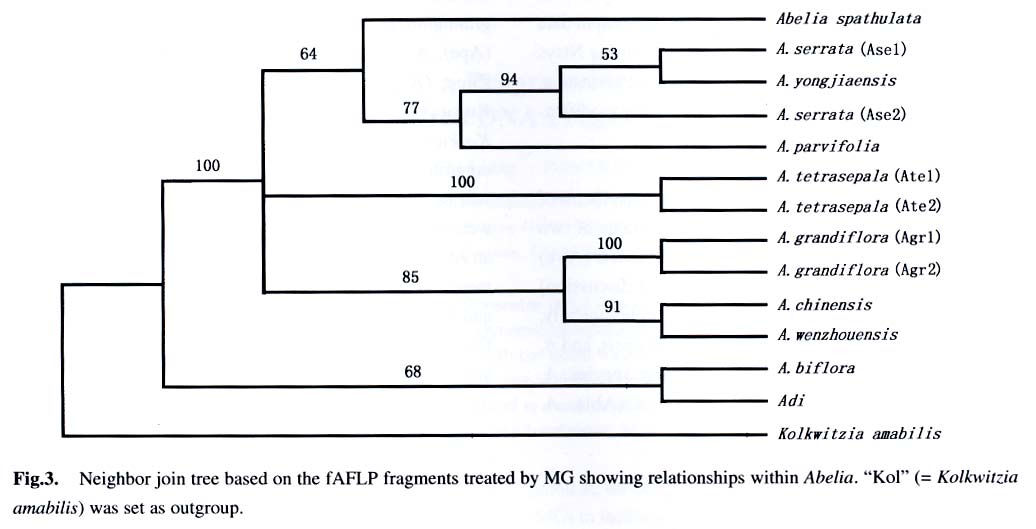
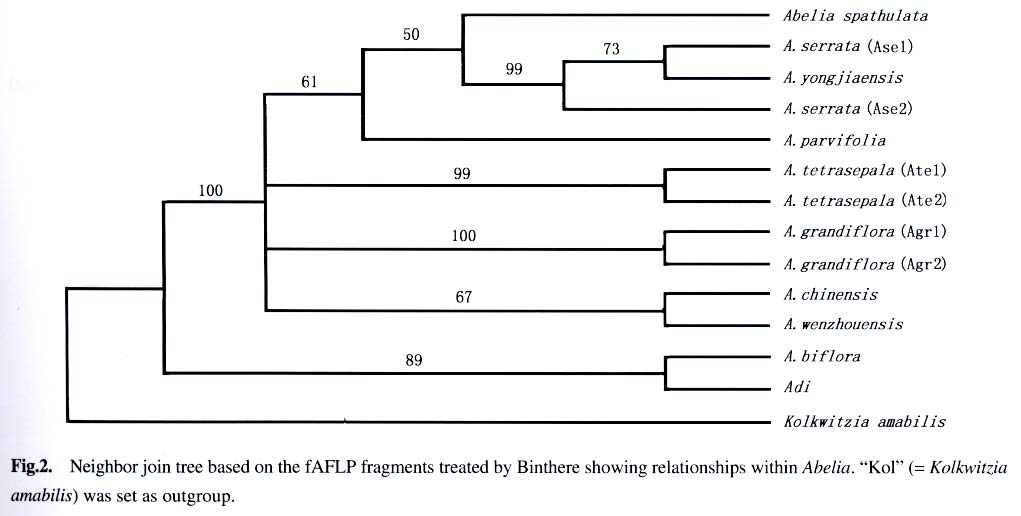
Sciences, Beijing 100093, China; Received: 2003-03-10 Accepted: 2003-04-10 Code Number: as03003 ABSTRACT A computer program called Matrix Generator (MG) was developed for transforming sized DNA fragments into a presence/absence data matrix. Dynamic computation was run to avoid errors introduced using fixed-bin-width arithmetic. MG can be used with bin sized fragments from AFLP, ISSR, RAPD, RFLP, and other molecular markers. The accuracy of MG was tested using fAFLP data of Abelia and the results show that MG results in higher resolution of taxa and is more reliable than programs of the similar usage. Key words: Abelia ; AFLP; computer software; data treatment; Matrix Generator (MG) A feature of population genetic studies is the sampling of large numbers of individuals from many populations and the screening of large numbers of loci. The sample size of a population should be large enough to allow statistical analysis; the number of populations should be reasonable to represent the variations of the species in question; and the number of loci should be high enough to give sufficient representative of the genome. Large sample sizes cause difficulties for manual manipulation, hence computer-aided raw data treatment is needed. Generally digital gel pictures are now collected using CCD, and DNA fragments can be readily sized using software such as Quantity One (BIORAD) or GeneScan (ABI). From an evolutionary viewpoint, DNA fragment data can only be useful when they are transformed into comparative data set. The software evaluated by us (Quantity One or GeneScan) to size the fragments turns out to be either too complicate to use or not very powerful at this phase of data assembly. Other programs, e.g., Binthere (Garnhart, 2001), are aimed at a specific purpose, i.e., to extract data directly from the sample files generated by GeneScan. This program is therefore convenient for fAFLP or fSSR, but not for RAPD and ISSR. Programs developed with the limitation of fixed bin-widths have the problem of binning two fragments of minor size difference into different bins. For example, two fragments of 49.95 bp and 50.05 bp might be mistakenly binned in bin 49-50 bp and 50-51 bp respectively when bin width was set 1 bp. To solve this problem, we developed a program named Matrix Generator (MG) to facilitate data treatment and make a matrix suitable to be analyzed by other programs after simple edition. The principle of MG is the axiom that size difference of the homologous fragments is due to experimental conditions (“resolution” in our program), therefore should be binned into the same site. Fragments with the least size differences are more likely to be homogenous, and therefore are first to be binned. With fixed bin width method, it is possible that two fragments are merged into the same bin, not because of the small difference of the fragment size, but the fragment sizes themselves. Therefore, MG does not use fixed bin width. Instead, the number of “bins” (“sites” in our program) depends completely on the materials in question and the gel used to resolve the fragments. “Resolution” here is analogous (but different) to bin width. Dynamic computation procedures start with the least different fragments and end when size difference between any two fragments reaches the value of resolution (i.e. large enough for the gel to separate them). This method minimizes the possibility of separating homogenous fragments from different sites or enclosing non-homogenous fragments into the same site. The first version of MG (Microsoft DOS version 1.0) was written simply for its ease and simplicity to generate a correct data matrix. MG bridges over the gap between data extracting software (such as GeneScan) and professional programs (such as Paup). For example, fragments can be accurately sized using Quantity One or GeneScan. Then the output data file serves as the input file of MG directly or with some minor editing using Microsoft Excel. The output data matrix of MG is suitable to be further analyzed using Ntsys (Rohlf, 1993), Paup (Sworfford, 1998), Phylip(Felsenstein, 2001), Rapdfst (Black, 1995), etc., after minor modifications to fit the formats of these programs. An Example fAFLP analysis was carried out on the genus Abelia of Caprifoliaceae to determine the taxonomic status of two collections (tentatively named as A. wenzhouensis (Awe) and A. yongjiaensis (Ayo) only for the ease of discussion) from Wenzhou City, Zhejiang Province. Morphologically, A. wenzhouensis is somewhat close to A. chinensis, and A. yongjiaensis is very similar to the Japanese species, A. serrata (Ase). Samples of A. biflora Turcz. (Abi), A. chinensis R. Br. (Ach), A. dielsii (Graebn.) Rehd. (Adi), A. grandiflora Hort. ex L. H. Bailey (Agr), A. parvifolia Hemsl. (Apa), A. serrata Sieb. and Zucc., A. spathulata Sieb. and Zucc. (Asp), and A. tetrasepala (Koidz.) Hara and Kurosawa (Ate), were collected from China and Japan. Kolkwitzia amabilis Graebn. (Kol) was used as an outgroup. Details of materials and experimental procedures will be described in a later publication. AFLP fragments were fluorescent dye-labeled, separated using PAGE with an ABI 377 DNA Sequencer, and sized by GeneScan. Fragments were binned using both Binthere and MG. Binthere and MG gave 295 and 210 informative sites, respectively. Distance matrices based on each method were compared and subject to Mantel test (Mantel, 1967) using programs incorporated in Nysys-pc. The two matrices correlated well (r = 0.941 63) though considerable differences existed between them (Fig.1). The data sets were further analyzed using Paup. Two neighbor-join trees were computed based on the same distance measure (Which one?). Figure 2 is the outcome of Binthere, and Fig.3 is that of MG. Both figures showed that A. wenzhouensis is clearly allied to A. chinensis. However, the kinship within A. serrata is different. Figure 3 seems more reliable because the Japanese sample “Ase1” and “Ase2” are likely to be closer each other than to the Chinese sample “Ayo”. Moreover, the resolution of Fig.2 is not as high as that shown in Fig. 3, i.e., the relationship between clusters “Agr” and “Ach-Awe” was unresolved. “Agr” was believed to be of hybrid origin and “Ach”was one of its parents (Rehder,1913). So, the relationship shown in Fig.3 seems more reasonable than that in Fig. 2. That is to say that data treated by MG seem to be more reliable. ACKNOWLEDGEMENTS The authors thank Tsuneo Funamoto, Biological Institute, Showa College of Pharmaceutical Sciences, for collecting materials in Japan; and Anthony Mitchell, Department of Botany, Field Museum of Natural History, for correcting the manuscript and suggestions for revision. REFERENCES
(Managing editor: HE Ping) Copyright 2003 - Acta Botanica Sinica. Free, full-text also available from http://www.chineseplantscience.com The following images related to this document are available:Photo images[as03003f3.jpg] [as03003f2.jpg] [as03003f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}