 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
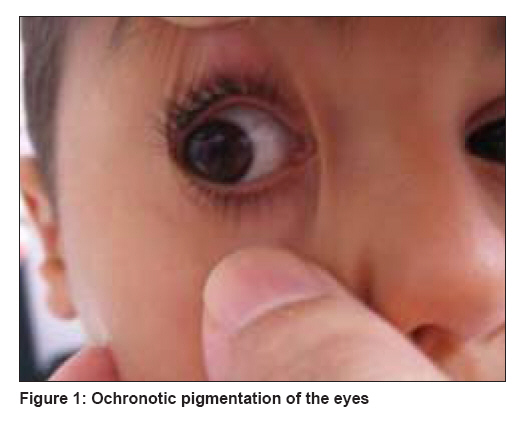
Indian Journal of Dermatology, Venereology and Leprology, Vol. 74, No. 6, November-December, 2008, pp. 700 Net Case From darkening urine to early diagnosis of alkaptonuria Peker Erdal, Yonden Zafer, Sogut Sadik Mustafa Kemal University, Medical Faculty, Hatay Code Number: dv08290 Abstract Alkaptonuria is a rare disorder of metabolism characterized by deficiency of homogentisic acid oxidase. Characteristic features include darkening of urine, ochronosis, and arthropathy. Darkening of urine is the only sign of the disorder in the pediatric age group, and it occurs at very early stage of the disorder, as reported by the parents. A 4-year-old boy presented to our clinic with the complaint of dark urine and bluish black staining of clothes. This darkening pointed to a positive physical history of bluish discoloration of sclerae which occurred off and on. We initiated treatment with ascorbic acid and a protein diet with restriction of phenylalanine and tyrosine (1.6 g/kg/d). This case report is significant because of the early diagnosis made.Keywords: Alkaptonuria, blue sclerae, dark urine, homogentisic acid Introduction Alkaptonuria is a rare (incidence, about 1 in 250,000) autosomal recessive disorder caused by deficiency of homogentisic acid oxidase. It is one of the first described metabolic disorders causing large amounts of homogentisic acid to accumulate in the body and then to be excreted in the urine. It is most common in the Dominican Republic and Slovakia. They excrete dark urine because of homogentisic aciduria that results from homogentisic acid oxidase deficiency. Biochemically, homogentisic acid is identified by paper chromatography. [1] Characteristic clinical features are darkening of the urine, ochronosis, osteoarthritis, and painful attacks of the large joints resembling acute inflammatory episodes. [2] The disorder is commonly seen in adulthood; but is also seen in childhood, though very rarely. In this report, we present a 4-year-old boy with alkaptonuria having ochronotic pigment deposited in the sclera and dark urine.Case Report A 4-year-old boy who was born at term with age-appropriate anthropometric parameters presented to our hospital with the complaints of darkening of the urine and bluish black staining of his underwear for the last 10 months. Both sclerae showed bluish black discoloration [Figure - 1]. The conjunctiva and cornea were normal, and no other abnormality was found in the eyes by the ophthalmologist. There was no discoloration of any other area of the skin. Ear cartilages were of normal color and in normal state. There was no joint pain, swelling, or deformity. X-rays of the hips, spine, and knees were normal. There were no cardiac signs or symptoms and ECG and echocardiogram were normal. There was no growth retardation. There was no history of any drug intake, and there was no family history of the same. Fresh urine of the child was normal in color, but its color changed to gray-black within a period of 1 hour or 2 hours. Addition of sodium hydroxide to the urine gave a typical black color [Figure - 2]. Laboratory data revealed normal blood tyrosine and urine tyrosine levels. Findings of the routine urinalysis were normal. Liquid gas chromatography revealed the presence of high amount of homogentisic acid in the urine. A diagnosis of alkaptonuria was made on the basis of the bluish black discoloration of the sclera, black-colored urine upon standing, and detection of homogentisic acid in the urine. Combination of oral ascorbic acid (1000 mg/d) twice daily and a low-protein diet (1.6g/kg/d) planned by a dietician was initiated as treatment for the patient. The patient is being followed up. Discussion Clinical manifestations of the disease include ochronosis and arthritis. These findings may not become evident until the third, fourth, or later decades of life. Darkening of urine is the only sign of the disorder in the pediatric age group, and it occurs at very early stage of the disorder, as reported by the parents. It is due to oxidation and polymerization of the homogentisic acid and is enhanced with alkaline pH. For this reason, acid urine may not darken after many hours of standing. This is one of the reasons why darkening of the urine may never be noted in an affected patient. In such cases, the diagnosis may be delayed until adulthood when arthritis or ochronosis occurs. This is because fresh urine looks normal in alcaptonuric patients. [3] Ochronosis is used to describe the darkening of tissue due to a slow accumulation of the black polymer of homogentisic acid in cartilage and other mesenchymal tissues, which is seen in the fourth decade of life. It is manifested clinically as dark, blackened spots on the sclera or as particularly diffuse blackish pigmentation of the conjunctiva, cornea, and cartilage. Arthropathy may cause disabilities but the life expectancy is usually normal. It involves the large joints (spine, hip, and knee) and is usually more severe in male patients and shows clinical symptoms resembling rheumatoid arthritis, but the radiologic findings are typically of osteoarthritis.[2] The characteristic manifestations of spinal abnormalities include widespread disk calcification, disk space narrowing, vertebral osteoporosis, and mild osteophytosis. The pathogenesis of arthritic changes is unclear. High incidences of cardiovascular involvement including pigment deposition can be intracellular, usually in the endocardium; or extracellular in the aortic valve. Other sites of deposition are the mitral and pulmonary valves, mitral annulus, mural endocardium, and areas of replacement fibrosis. [4] Ocular pigmentation is especially prominent and appears in approximately 70% of the patients. [5] Referred to as Osler′s sign [Figure - 1],[Figure - 2], ochronotic pigment deposition is confined to the exposed area of the sclera and becomes evident during the third decade of life. [6] In our case, early occurrence of this sign was interesting. For this reason, we presented it. The diagnosis is confirmed by measurement of homogentisic acid in urine. Affected patients may excrete as much as 4 to 8 g of this compound daily. The inability to convert homogentisic acid to maleylacetoacetic acid results in accumulation of homogentisic acid and a product of its oxidation, benzoquinone, which induces tissue injury. [7] Homogentisic acid is a strong reducing agent that produces a positive reaction with ′fehling′ or ′benedict′ reagent but not with glucose oxidase. The enzyme is produced only in the liver and kidney. The gene for alkaptonuria was mapped to the long arm of chromosome 3, and several disease-causing mutations were identified. [8] Diagnosis in our case was made on the basis of the presence of the triad of ochronotic pigmentation of sclera, urine which turns black upon alkalization, and detection of homogentisic acid in the urine. There is no effective treatment and no definite therapeutic protocol for this disease, but ascorbic acid, low-protein diet with restriction of phenylalanine and tyrosine, NTBC (nitisonine), or their combined therapy are the different modalities of treatment. Different clinical trials analyzed the effectiveness of a low-protein diet and ascorbic acid treatment. [9],[10],[11] Morava et al. [12] reported improvement in clinical symptoms and reversal of radiological evidence of joint involvement in a pubertal alcaptonuric patient receiving mild protein-restriction therapy and ascorbic acid. We also used combined therapy with ascorbic acid and protein restriction in our patient. Nitisinone inhibits homogentisate production; however, the long-term efficacy and side effects of such therapy are unknown. [10] References
Copyright 2008 - Indian Journal of Dermatology, Venereology and Leprology The following images related to this document are available:Photo images[dv08290f1.jpg] [dv08290f2.jpg] |
| |||||||||


{kind=link}
{kind=link}