 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
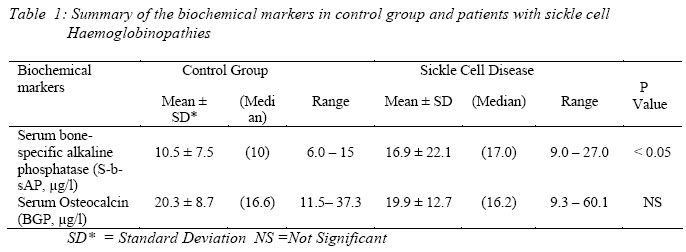
Nigerian Journal of Physiological Sciences, Vol. 21, No. 1-2, 2006, pp. 21-25 OSTEOCALCIN AND BONE-SPECIFIC ALKALINE PHOSPHATASE IN SICKLE CELL HAEMOGLOBINOPATHIES E. C. AZINGE and D. M. BOLARIN*, Department of Clinical Pathology, College of Medicine, University of Lagos, Lagos, Nigeria. *Department of Chemical Pathology, College of Medical Sciences, University of Calabar, Calabar, Nigeria. Received: 15/9/06Accepted: 16/11/06 Code Number: np06005 Summary: Osteocalcin or bone gamma-carboxyglutamic acid (gla) protein and Bone-specific alkaline phosphatase (b-AP) total protein levels were evaluated as indicators of bone turnover in twenty patients with sickle cell haemoglobinopathies and in twenty normal healthy individuals. The serum bone-specific alkaline phosphatase total protein level was measured by immunoradiometric (IRMA) method. The concentrations of serum bone-specific alkaline phosphatase total protein were higher in the study group than in the control group (p < 0.05). The serum osteocalcin (BGP) showed no significant difference with the control healthy subjects. There was no correlation between the serum osteocalcin and serum bone-specific alkaline phosphatase total protein in the patient group. In conclusion, serum bone-specific alkaline phosphatase total protein determined or measured by IRMA can be considered a sensitive marker of bone turnover and could be especially useful as valuable non-invasive biochemical marker for identifying sickle cell patients with bone complications. Key Words: Bone-specific alkaline phosphatase (b-AP); Osteocalcin; sickle cell haemoglobinopathies. Introduction Sickle cell haemoglobinopathies are probably the commonest known hereditary blood disorders in Nigeria (David et al, 1993; Ballas, 1995). The sickle cell anaemia (SS) is the homozygous state in which the sickle gene is inherited from the father and the mother. Sickle cell disease may also occur in a heterozygous form in conjunction with other haemoglobin beta chair abnormalities. The common heterozygous forms include sickle haemoglobin C disease (HbSC), and sickle cell haemoglobin thalassaemia (HbSβ+thal. or HbSβ°thal.) (David et al, 1993; Ballas, 1995; Ballas 2001; Smith, 1996; Aster, 2004; Iwegbu and Fleming 1985). Sickle cell disorder may be classified as an uncompensated haemolytic anaemia in which the life span of the erythrocytes is shortened due to increased rate of destruction and this is insufficiently balanced by increase in erythropoiesis. Vascular occlusion plays a major role in the clinical course of sickle cell disorders (Lee et al, 1981; Embury et al, 1994; Francis and Johnson, 1991; Hebbel, 1991; Powars, 1990). Vascular occlusion may occur in both the microcirculation and the macrocirculation. When vascular occlusion occurs in the microcirculation it leads to acute sickle cell painful episodes, which is the hallmark of the sickle cell disorder. Vascular occlusion in the macrocirculation is associated organ failure (Lee et al, 1981; Embury et al, 1994; Francis and Johnson, 1991; Hebbel, 1991; Powars, 1990). Patients with sickle cell disorders often suffer from both chronic haemolytic anaemia, which causes bone marrow hyperplasia (Lee et al, 1981; Embury et al, 1994; Francis and Johnson, 1991; Hebbel, 1991; Powars, 1990) and repeated episodes of vaso-occlusion (Lee et al, 1981; Embury et al, 1994; Francis and Johnson, 1991; Hebbel, 1991; Powars, 1990). The results of infection and infarction, which lead to orthopaedic complications, have been well documented by several authors (Lee et al, 1981; Embury et al, 1994; Francis and Johnson, 1991; Hebbel, 1991; Powars, 1990). Avascular necrosis is the commonest and a major chronic complication of sickle cell disorders (David et al, 1993; Ballas, 1995; Ballas 2001; Smith, 1996; Mitchell et al, 1986; Rao et al, 1985; Rao et al, 1989; Amundsen et al, 1984; Milner et al, 1993; Mitchell et al 1987). The sickle cell disease is a cause of hip deformities in the patients (David et al, 1993; Ballas, 1995; Ballas 2001; Smith, 1996; Lee, et al, 1981; Embury et al, 1994; Francis and Johnson, 1991; Hebbel, 1991; Powars, 1990). Involvement of the humeral head is also very common (Rao et al, 1985; Rao et al, 1989; Amundsen et al, 1984; Milner et al, 1993; Mitchell et al, 1987). More than 30 percent of patients with sickle cell disorders end up with complications of avascular necrosis of the bone (David et al, 1993; Ballas, 1995; Ballas 2001; Smith, 1996). The disappointing results of treatment of avascular necrosis after bony collapse have led to the suggestion by many workers that future efforts should be directed to the early detection or diagnosis of this bone disease before the onset of the skeletal or structural failure in patients with sickle cell haemoglobinopathies. Bone scan with technetium 99mTc and magnetic resonance imaging (MRI) can detect disease but these tests are very expensive and difficult to use routinely in a developing country like Nigeria (David et al, 1993; Ballas, 1995; Ballas 2001; Smith, 1996; Rao et al, 1985; Rao et al, 1989; Amundsen et al, 1984; Milner et al, 1993; Mitchell et al, 1987). Recently there has been introduction of new biochemical markers of bone metabolism such as osteocalcin and others (Bettica and Moro, 1995; Kleerekoper 1997; De la Piedra et al, 1996; Bolarin, 1996). a. Osteocalcin Osteocalcin or bone gammacarboxyglutamic acid (gla) protein (BGP) is the most abundant non-collagenous protein found in bone (Bolarin, 1996). Osteocalcin has a molecular weight of 5.7 kD. It is synthesized exclusively by osteoblasts and by its dental form, the odontoblasts (Lian and Grundberg 1988; Hauschka et al, 1989). Osteocalcin represents the activity of osteoblasts, since it is synthesized and released into circulation by the osteoblasts. The protein is eliminated by glomerular filtration and degraded by the renal tubule (Bolarin, 1996; Lian and Grundberg 1988; Hauschka et al, 1989; Garnero et al, 1994). Measurement of osteocalcin is by immunoassays using monoclonal or polyclonal antibodies. It is advised that blood samples be collected in ice and plasma or serum is stored at –20 oC or –70 oC. It should be thawed once to prevent degradation of the protein (Bolarin, 1996; Lian and Grundberg 1988; Hauschka et al, 1989; Garnero et al, 1994). b. Alkaline phosphatase (EC. 3.1.3.1) Bone-specific alkaline phosphatase (b-AP) is a plasma membrane enzyme. For many years this enzyme has been the most commonly used biochemical marker to assess osteoblast function (Bolarin, 1996). Bone-specific alkaline phosphatase activity correlates with bone formation rate in metabolic disease of the bone (Bolarin, 1996; Grundberg, 1993; Bolarin, 2001) and during normal physiological bone growth (Bolarin, 1996; Grundberg, 1993; Bolarin, 2001). The serum alkaline phosphatase activity is present in tissues other than bone. The isoenzymes can be divided into four groups (placental, carcinoplacental, intestinal, and hepatic, renal, skeletal group). The major fraction of the total serum is from the liver and bone isoforms or isoenzymes, which are both products of the same genetic loci (Bolarin, 1996; Grundberg, 1993; Bolarin, 2001). It is often difficult to distinguish between the two isoenzymes. But distinction is necessary for clinical use (Bolarin, 1996). There are several methods for the estimation of serum bone-specific alkaline phosphatase. These methods include inactivation some of the isoenzymes by heat or using chemical compounds, precipitation with wheat germ lectin and separation by electrophoresis. These techniques, however, do not seem to provide the sensitivity, specificity and reliability required for routine clinical use (Bolarin, 1996; Grundberg, 1993; Bolarin, 2001). A two-site immunoradiometric assay that preferentially recognizes bone-specific alkaline phosphatase is generally being used in most clinical or research laboratories (Bolarin, 1996; Grundberg, 1993; Bolarin, 2001). These new assay methods have shown improved diagnostic accuracy with regards to bone diseases and their therapeutic monitoring (Bolarin, 1996; Grundberg, 1993; Bolarin, 2001). In the present preliminary study, serum osteocalcin is assayed by enzyme immunoassay using monoclonal antibody. Serum bone-specific alkaline phosphatase protein (b-AP) was also measured by immunoradiometric assay (IRMA). Both biochemical markers were used to investigate bone turnover in patients with sickle cell haemoglobinopathies. Materials And MethodsStudy Population Twenty patients consisted of 9 men and 11 women with sickle cell haemoglobinopathies. Mean age ± standard deviation was 31.6 ± 11.5 years (median 30 years, range 18 -5 1 years). A reference control group of 20 apparently healthy subjects (9 males and 11 females), age range 20 – 52 years (median 30 years). None of the persons had a history serious disease or was taking drugs known to affect bone metabolism or the blood vessels. The control had no history of fractures nor any nutritional, metabolic, liver, renal or bone disease. None of the females were on contraceptives prior to obtaining blood samples. All control subjects had haemoglobin AA. The control groups were individuals of African-American ancestry just like the patients used in the study. The diagnosis of sickle cell disease was based on the haemoglobin pattern on cellulose acetate electrophoresis. Family studies were consistent where available. The genotypes of the patients were divided as follows:
Five patients were identified clinically and by physical diagnostic methods (such as radiography, etc.) to have skeletal complications due to sickle cell disease. All the other patients were asymptomatic for skeletal complications at the time blood samples were taken for the biochemical assays. The clinical history and routine liver function tests and urinalysis carried out on the experimental subjects including the control group showed that none of the patients or control subjects had liver, renal, or inflammatory disease during the time blood and urine samples were taken for the assays. The patients were attendees at sickle cell disease out-patient clinic. Blood samples: Blood samples were collected at the same time in the morning during clinic. Sera were separated from blood by centrifugation at 4 °C and were stored frozen at -60 °C until assayed. Assays were carried out within 1 to 2 weeks of storage. Assay of Biochemical markers of bone metabolism a. Determination of serum osteocalcin (SBGP): Serum levels of osteocalcin (S-BGP) were determined in duplicate samples using an IRMA kit from CIS bio International, (GifSurYvette, France). The tracer is a 125Imonoclonal antibody against human BGP. The monoclonal antibody used in this study is raised against human osteocalcin and therefore measured total serum osteocalcin protein levels. The serum concentration of osteocalcin was expressed as μg/L. b. Determination of serum bone-specific alkaline phosphatase (S-b-AP): Serum b-AP protein level was determined with an IRMA (Tandem-R-OSTASE, Hybritech, San Diego, CA), using two monoclonal antibodies, specific for b-AP over liver alkaline phosphatase. Briefly, 100 µl of standard or serum and 100 µl tracer (100 nCi [125I] anti-b-AP mouse monoclonal antibody) were added to beads coated with mouse monoclonal antibody directed to human b-AP. After incubation for 19 ± 2 h at 4 oC, the solid beads were washed three times with detergent solution. Total and bound radioactivity was counted in a gamma-counter. The level of b-AP protein is expressed as µg per liter. Statistical Analysis The results are presented as means ± SD. Median values and range are included. The mean ± SD are compared, as applicable, by Student’s t-test. ResultsThere were no statistically significant differences in age between the groups. The results of the biochemical markers of bone formation, serum osteocalcin (S-BGP) and serum bone-specific alkaline phosphatase (b-AP) are summarized in Table 1. The mean level of serum osteocalcin was not statistically (p < 0.9) different from the control group. The concentration of bone-specific alkaline phosphatase protein was significantly higher in the patients with sickle cell disease than in control group (p < 0.05). The upper limit of the reference range (i.e. mean + 2 standard deviation, SD) of the control for the bone-specific alkaline phosphatase was 25.5 µg/l. The upper limit of the reference ranges for serum osteocalcin (S-BGP) were exceeded by 2/20 patients or 10 per cent and 11/20 patients or 55 per cent of all the sickle cell disease patients studied respectively. DiscussionSerum alkaline phosphatase is used extensively for the assessment of bone turnover in patients with various metabolic bone disorders, but its clinical use is often impaired by serum changes occurring in non-bone alkaline phosphatase. In this study, we used the two-site immunoradiometric assay (IRMA) developed by Hybritech (Tandem-R Ostase) to determine the bone isoenzyme in sera of normal subjects and in patients with sickle cell haemoglobinopathies. With this IRMA, serum bone-specific alkaline phosphatase protein increased significantly in patients with sickle cell disease when compared with those in the normal healthy subject group. The results of bone-specific alkaline phosphatase total protein levels in this study agree with data obtained in patients with metabolic bone disease, showing increased serum bone-specific alkaline phosphatase (Duda et al, 1988). Our data shows that serum bone-specific alkaline phosphatase total protein determination may be useful in early diagnosis of bone changes in sickle cell haemoglobinopathies, without any sign of hepatic disorder (Bolarin, 1983). There was no difference in the mean values of serum osteocalcin in both control group and patients with sickle cell haemoglobinopathies. The value of osteocalcin as a biochemical marker of bone matrix formation is unclear as fluctuation in circulating osteocalcin may only reflect changes in the equilibrium between bone matrix and bone (Bolarin, 1996; Duggan, 2001). Osteocalcin is unstable in circulation and subject to variable vitamin K-dependent carboxylation. Because of the heterogeneity of circulating osteocalcin, different assays give differing results (Bolarin, 1996; Duggan, 2001). In conclusion, we have shown that IRMA assay of serum bone-specific alkaline phosphatase total protein was more sensitive and specific than the determination of serum osteocalcin (S-BGP) to detect the increase in bone turnover due to bone remodeling in sickle cell haemoglobinopathies. The pilot data presented here also form the basis for further confirmatory studies leading to development of noninvasive biochemical markers to monitor skeletal complications in sickle cell disease. It may be useful to monitor the course of therapy of these complications in these patients, since it is characterized by increased sensitivity and organ specificity (Behr and Barnett, 1986). AcknowledgmentsThis study was carried out while one of the authors, Dr. Bolarin was working in Wayne State University, Detroit, Michigan, USA and it was supported by the University Interdisciplinary Grant 1444981. The authors are grateful to Dr P. Swerdlow, Division of Haematology for the patients. References
© Physiological Society of Nigeria 2006 The following images related to this document are available:Photo images[np06005t1.jpg] |
| |||||||||

{kind=link}