 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Iranian Journal of Pediatrics, Vol. 18, No. 1, March, 2008, pp. 62-66 Case Report Congenital Embryonal Rhabdomyosarcoma with Prenatal Onset Fatemeh Khatami*1, MD, Neonatologist; Ahmad Bazrafshan2,MD, Pediatric surgeon; Maryam Monajemzadeh3, MD, Clinical Pathologist; Masood Seyed1, MD, Pediatrician 1Department of
Pediatrics, Tehran University of Medical Sciences, Tehran, IR Iran Received: 14/05/07; Revised: 22/07/07; Accepted: 14/08/07 Code Number: pe08010 Abstract Objective:Rhabdomyosarcoma (RMS) is the
single most common type of soft tissue sarcoma in children and adolescents but
it is extraordinarily rare in neonates. Extremity RMS comprises 20% of all
sites, occurs more commonly in the leg than in the arm and accounts for 9% of
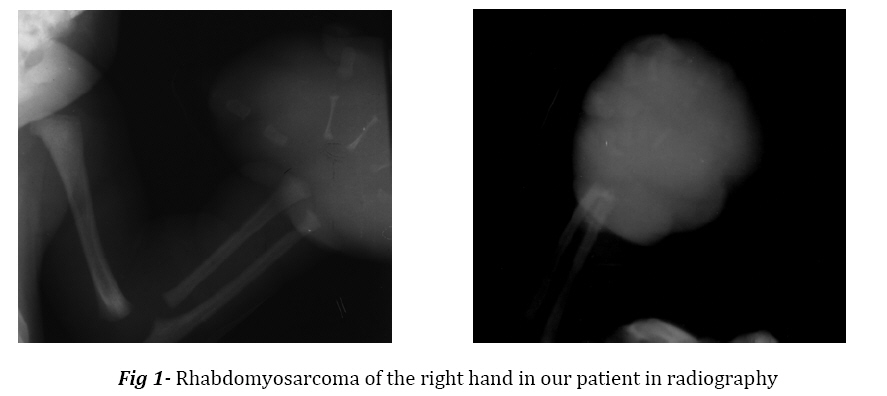
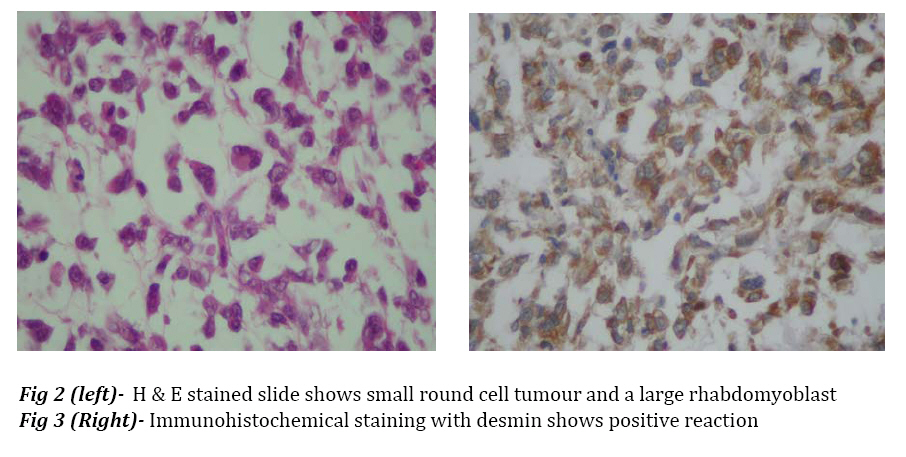
all RMS cases. Key Words:Congenital, Rhabdomyosarcoma, Embryonal, Antenatal Introduction Rhabdomyosarcoma (RMS) is a soft tissue tumor originating from immature mesenchymal cells, and accounts for about one-half of soft tissue sarcomas in children, but it is rare in neonate and little is known about it in this age period[1-6]. The four major pathologic subtypes are outlined by the international classification of RMS: 1) botryoid and spindle cell RMS, with most favorable prognosis (variant of embryonal); 2) embryonal (ERMS), with intermediate prognosis; 3) alveolar (ARMS), with poor prognosis; and 4) undifferentiated sarcoma, also with poor prognosis. Embryonal RMS belongs to the class of small round blue cell tumors. The ERMS make up about 60-70% of RMS cases[6,7]. RMS may be present at birth, 1-2% of all cases are congenital with intrauterine origin[8]; neonatal tumors can be diagnosed at birth or in the first 28 days. There is no racial predilection; the tumor is slightly more common in boys. Some prenatal risk factors have been found for development of RMS. A number of inherited syndromes and genetic diseases demonstrate predisposition for developing RMS[6,9]. Approximately one third of RMS cases are associated with at least one congenital anomaly[4,7,10,11]. Chromosomal abnormality, loss of heterozygosity at the 11p15 locus is known in ERMS[6,7,9,12,13]. Approximately one half and three-fourths of the sarcomas of the extremities are alveolar[6,7] and occur more commonly in the leg, so this case is unusual form of embryonal histological subtype in extremity. Case Report The patient, a term newborn boy, was born to 25-year-old, gravida 4, Para 3, mother. The prenatal ultrasonography showed a mass on the right hand of the fetus, otherwise prenatal history was unremarkable. Family history for drugs, malignancy, and hereditary diseases was negative. The baby was delivered vaginally; apgar score at birth was 7 and 10 at one and five minutes respectively. Birth weight 3 kg, length 52 cm and head circumference 35 cm. Physical examination was normal except for right hand with a huge mass (6×9 cm) in the right palm of the hand, encroaching the tip of the fingers. The mass was pink, soft, nontender, lobulate, heterogeneous, and fixed to the surrounding tissue. It was covered with very tinny skin with dilated and tortuous blood vessels. The tumor completely filled the right hand. There was palpable regional lymph node (1×1 cm) in ipsilateral subaxillar region. In the first hour of life he was admitted to neonatal intensive care unit for evaluation. He was otherwise in good health. He underwent radiological and ultra sonographic work up, the laboratory work up included complete blood count, urinalysis, electrolytes, blood urea nitrogen, creatinin level, liver function tests, bone marrow aspiration and lumbar puncture. The molecular biologic studies were not available. There was no abnormal finding except for imaging, which showed a soft tissue mass in the right hand; it had affected metacarpal and phalangial bones (Fig 1). Histological examination of incision biopsy (0.8 cm tissue sample) showed a tissue with small round cell tumor. Tumor node metastasis (TNM) pre-treatment staging was T2bN1M0. The tumor was fixed and unresectable; therefore, on day 12 of life the patient underwent surgery for amputation of right hand 4 cm over the wrist and regional lymph node excision. Gross morphologic description of the tumor after surgery was a soft and elastic mass, 6×6×9 cm, with skin transparency and white color appearance of underlying tissue. Histological examination showed a neoplasm composed of round, oval and polygonal cells arranged in nondescript sheet and focal fascicle formations. In the areas of edematous and myxoid stroma neoplastic cells were scattered. Rare rhabdomyoblasts with eosinophilic cytoplasm and also tadpole cells were identified. Nuclei were inconspicuous mostly but nuclear pleomorphism was seen here and there and also several atypical mitotic figures and bizarre nuclear shapes were seen (Fig 2). Immunohistochemical study showed positive reaction with desmin, CD99, actin and myogenin, confirming the diagnosis of RMS (Fig 3). Regional lymph nodes were also involved by neoplasm and necrotic areas. The patient was discharged on 20th day of life, receiving systemic chemotherapy, but Radiotherapy was not applied. The patient was readmitted at the age of 28 days for respiratory distress, fever and seizures. Physical examination revealed hepatomegaly, anemia, right subaxillar lymphadenopathy, mouth thrush, tachypnea and tachycardia, leukopenia, and neutropenia. There was also renal dysfunction. He died at 29th day of life. Discussion Extremity RMS comprises 20% of all sites[6,7], occurs more commonly in the leg than in the arm and accounts for 9% of all RMS[7]. Up to one half of patients with extremity tumors have regional lymph node involvement. Tumors at these sites are predominantly of unfavorable alveolar histological pattern and the highest relapse and lowest survival rate.[6,7,14] Brain metastasis develops in approximately 2.4% of patients with ERMS or ARMS. It is interesting to note that this incidence rate is even higher for patients with RMS of the extremities.[3] Owing to strong tendency towards early nodal metastasis, nodal sampling techniques should be employed.[7] In one report, during 20 years, of 50 children with RMS aged less than l year at diagnosis, 15 were considered as having congenital RMS[8]. Rodriguez et al, reported four patients with neonatal RMS treated during 37 years (1962-1999), one patient had embryonal and the other three alveolar RMS with skin and brain metastases; diagnosis was made at the age of at 2-7 weeks. In none of them the hand was involved. In two other reports of neonatal alveolar RMS there was only one case with forearm involvement[3]. Lobe et al reported 3217 eligible patients, 14 were less than 30 days old at the time of diagnosis. Two thirds of tumors were Embryonal.[15] Prenatal diagnosis by magnetic resonance imaging or ultrasound can be useful. Skelton and Goodwin reported a neonate with intra oral embryonal RMS that was diagnosed on antenatal ultrasound scan.[16] As far as we know this is the second published case of RMS with congenital in nature, antenatal feature, and postnatal progressive clinical course of hand tumor with embryonal histological subtype and unfavorable prognosis. The findings suggest that the malignant cell of neonatal patients with alveolar RMS is present during fetal development and probably before the blood brain barrier is developed[3,10]. The studies of Skelton and ourselves suggest that the above findings may be pathologic course in subtypepatients with ERMS as the same as alveolar subtype. The factors which probably affect the prognosis include immaturity of organs resulting in organ insufficiency during chemotherapy and low immunity in neonatal period, the presence or absence of distant metastasis, site, surgical resectability, histology, pre-treatment staging classification for intergroup RMS (IRS) or tumor, node, metastasis (TNM) and clinical group staging system[4,7,14,17]. According to Joshi et al. age is an independent prognostic factor[18]. Dillon et al. identified 32 neonates during 20 years (1971-1991) with soft tissue sarcoma, 11 cases being RMS and extremity involved in 9 cases. Survival rate as in Andrassy study was independent of pathology and extent of disease[19,20]. The presence of necrosis and small round cell configuration coincide with poor prognosis[15]. The presence of these last two findings in our patient with pre-treatment IRS stage 3 T2bN1M0 and probably prenatal onset were more likely cause of poor prognosis. Optimal therapy for neonatal RMS has not been well established[15], the excellent response to chemotherapy has allowed less aggressive surgery[21], amputation usually is not necessary[6] and was not primarily performed[14,22]; the use of radiotherapy is restricted by very high risk of side effects[8] and should be avoided in the newborns.[1,14,17] Conclusion Congenital embryonal rhabdomyosarcoma is a rare form of sarcomas with congenital in nature, .antenatal feature and post natal progressive clinical course of sarcomas of extremities in newborn infants. Acknowledgment The authors would like to thank Dr. Derhami for his kind assistance in pathologic diagnosis. References
© Copyright 2008 - TUMS PUBLICATIONS The following images related to this document are available:Photo images[pe08010f1.jpg] [pe08010f2-3.jpg] |
| |||||||||

{kind=link}
{kind=link}