 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
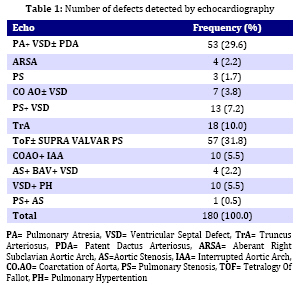
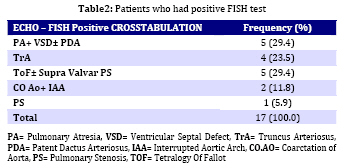
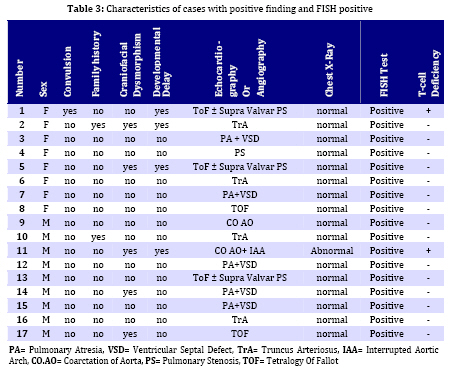
Iranian Journal of Pediatrics, Vol. 19, No. 1, March, 2009, pp. 11-17 Microdeletion Study in Children with Selective Congenital Heart Disease; an Iranian Multicenter Study Ali Akbar Zeinaloo1,2, MD; Abdorazaagh Kiani1, MD; Parvin Akbari-Asbagh1, MD; Mohammad-Reza Noori-Dalooi3, PhD; Elham Ghadami-Yazdi4,MD; Tayebeh Sabokbar5, MSc; Asgar Aghamohammadi1,2, MD; Mahmood-Gholam Alemohammad4, MD; Sima Rafeyan6, MD; Jila Dastan7,8, MSc, MD; Saeed-Reza Ghaffari3, 4,5,7,8 ,MSc, MD, PhD 1. Department of Pediatrics, Tehran University of Medical Sciences, Tehran, IR Iran Received: 17/01/08; Revised: 21/05/08; Accepted: 25/07/08 Code Number: pe09002 Abstract Objective: Determining the frequency of chromosome 22q11.2 microdeletion in children with congenital cardiac conotruncal abnormalities using Fluorescence in-situ Hybridization (FISH) technique and estimating relation between DiGeorge Syndrome and cardiac conotruncal abnormalities. Key Words: FISH; Congenital heart disease; Microdeletion of chromosome 22q11.2; DiGeorge syndrome Introduction Congenital heart disease (CHD) is the most common birth defect and the leading cause of mortality in the first year of life with a prevalence of 1% in live births and 10% in spontaneously aborted fetuses[1]. CHD is mainly a multifactorial disorder characterized by: a) 90% multifactor disorders, b) 8% chromosomal and single gene disorders and c) 2% environmental teratogens[2] in majority of cases, CHD occurs as an isolated malformation; however, up to 33% have associated anomalies[3]. Among chromosomal disorders, chromosome 22q11.2 micro-deletion, is the most common syndrome associated with CHD with estimated prevalence of approximately 1 in 4000 live births[4,5], 90% of which are sporadic[6,7]. The various conditions associated with chromosome 22q11.2 microdeletion include DiGeorge anomaly (DG), Velocardiofacial syndrome (VCFS), Conotruncal anomaly face syndrome, isolated conotruncal heart defects and Cayler cardiofacial syndrome[8-11]. As a result of their common genetic origin and phenotypic overlap, the syndromes were collectively referred to as “CATCH 22” (Cardiac abnormality, Abnormal facies, Thymic hyperplasia, and Cleft palate, Hypocalcemia and chromosome 22q11.2 microdeletion)[8-12]. The most common cardiovascular anomalies in patients with this syndrome include Tetralogy of Fallot (ToF), Interrupted Aortic Arch, Persistent Truncus Arteriosus and Ventricular Septal Defect (VSD), requiring early clinical intervention[13]. Several documented studies showed the presence of chromosome 22q11.2 microdeletion in patients with CHDs ranging (0%- 80%)[11,14,19]. This study investigates the frequency of Chromosome 22q11.2 microdeletion and T cell deficiency in Iranian young population with selective congenital heart disease. Subjects and Methods A total of 180 patients (106 males, 74 females) were enrolled in the study. The patients were selected among children with cardiac abnormalities (excluding those with known numerical chromosome aberrations) referred consecutively from Tehran University affiliated hospitals to the Departments of Pediatric Cardiology of the Children's Medical Center and Imam Khomeini Hospital over a four year period (2004-2007). Median age of the patients at the time of study was 18 months (The age ranged from 3 days to 16 years). All children received a general clinical pediatric examination together with cardiological and genetic evaluation by pediatric cardiologists and a clinical geneticist. The clinical data including weight, height, type of CHD, presence of dysmorphic features, extra cardiac malformations (if any) and psychomotor development werealso ascertained. Chromosomal analysis was then carried out on peripheral blood lymphocyte culture using standard protocol with suitable modifications (Sea bright 1971)[19]. G-banded metaphase chromosomes were then screened at 450 band level. The chromosomes were analyzed according to the International System for Human Cytogenetic Nomenclature (ISCN 1995)[20]. Peripheral venous blood of each patient was used for Fluorescence in Situ Hybridization (FISH) analysis as previously described[15,16]. Briefly, slides were prepared using interphase cells spotted on to cleaned microscope slide, then immersed in 2 x SSC (3 mol/l NaCl, 3 mol/l Na3-citrate), pH 7.0 for two minutes each and dehydrated in an ethanol series (70%, 85%, 100%) for two minutes each.Then Pre- and post-hybridization FISH was performed using specific probes (DiGeorge/VCFS TUPLE1 Cytocell) based on the protocol recommended by the manufacturer. FISH images were captured using a fluorescent microscope (Leica CW 4000) equipped with a CCD camera and FISH analysis software (Leica). Subsequently, patients with chromosome 22q11.2 microdeletion were studied for T cell abnormalities. Patients' white cell count, lymphocyte count, T cell numbers, flowcytometry and clinical history of infections were reviewed. Findings One hundred eighty patients withconotruncal cardiac abnormalities aged 3 days - 16 years (Median 18 months) including 106 (59%) males and 74 (41%) females, were enrolled in this study. The distribution of different congenital heart diseases among these patients is shown in Table 1. The most common congenital heart defect was Tetralogy of Fallot (ToF) accompanied with supravalvular pulmonary stenosis. FISH analysis revealed chromosome 22q microdeletion in 17 (9.5%) patients. Among the FISH positive patients, 5 (29.4%) ToF plus supravalvular pulmonary stenosis, 5 (29.4%) had Pulmonary Atresia (PA) with ventricular septal defect (VSD), 4 (23.5%) had truncus arteriosus (TA). More details are shown in Table 2. According to the clinical characteristics of the patients, in FISH positive group, 4 (23.5%) had developmental delay, 5 (29.6%) had craniofacial dysmorphism and two (12%) had positive family history. Nine out of 17 FISH positive patients were male and 8 female. The spectrum of heart defects, and other related findings in FISH positive patients are shown in Table 3. Among 17FISH positive patients, only 2 patients had mild thymic aplasia diagnostic of DiGeorge/VCFS syndrome. The remaining 15 patients had no evidence of diminished T cells. Discussion The present study was performed to investigate the 22q11.2 microdeletion among Iranian children with congenital heart disease (CHD). FISH analysis using TUPLE 1 (Cytocell) probe on all the 180 CHD patients involved in the study revealed microdeletion in 9.5% (17) of the cases. In a population-based study of 255849 births Lorenzo et al[21] identified 43 children with 22q11.2 deletion.The overall prevalence was 1 in 5950 births. They concluded that such population-based studies would be useful to medical professionals and policy makers in planning for the optimal care of people with the 22q11.2 deletion[21,22]. Tobias et al[23] studied on 551 children with cardiac anomalies using FISH analysis and revealed 12% of cases with 22q11.2 microdeletion. In another study McDonald-McGinnet al[24] investigated the phenotype of the 22q11.2 deletion individuals, 30 individuals with a 22q11.2 deletion were identified following the diagnosis in a relative. Sixty percent of the patients had no visceral anomalies; this showed the importance of deletion testing in parents of affected probands. The authors emphasized the importance of broadening the index of suspicion in order to provide appropriate recurrence risk assessment, cognitive remediation, and medicalmanagement. Studies by Fokstuen et al[26] with a sample size of 110 non-selective (syndromic+ non-syndromic) CHD using the D22S75 DiGeorge chromosome region probe (FISH), showed 9/51 (17.6%) syndromic patients. Five were of maternal origin and four of paternal origin. None of the 59 patients with isolated congenital cardiac defect had a 22q11.2 deletion. Borgmann et al[25]concluded that testing for the 22q11.2 microdeletion is clearly indicated in cases when even mild extracardiac abnormalities are present, particularly in very young infants. In another study carried out by Gawde et al[27] with a sample size of 105 prospective cases which included 6 families with isolated, non-syndromic cardiac defects, showed only 5.71% chromosome 22q11.2 microdeletion. They suggested that testing for chromosome 22q microdeletion in isolated non-syndromic patients using FISH technique is mandatory even when mild/unspecific extracardiac abnormalities are seen in the patients. At the present FISH study the microdeletion of 22q11.2 was detected in 9.5% of patients. Five out of 22 syndromic patients (DGS/VCFS indices) had 22q11 micro-deletion. Ryan et al[14] presented clinical data on 558 patients with deletions within the DiGeorge syndrome critical region of chromosome 22q11.2, and immunological problems were very uncommon. They reported only 4 major immune function abnormalities and 15 cases with clinical history of infection. Sullivan [28] reported low T-cell numbers in 75% to 80% of infants who had chromosome 22q11.2 deletion syndrome. In most cases this problem was mild or moderate and did not have an impact on the procedures. However, it is not known whether postoperative infections are increased in this population. Less than 1% of patients who have the deletion lack T cells, but these patients represent a special category at the time of cardiac surgery and require protection from infection and blood products. Blood products can induce graft-versus-host disease in patients without T cells, and graft-versus-host disease from transfusions almost always is fatal. At the present study 2% of all patients and 12% of patients with microdeletion 22q11.2 had mild to moderate T cell/thymus abnormality. Children who have chromosome 22q11.2 deletion syndrome and a mild to moderate decrement in the T-cell count have largely normal immunoglobulin levels and T-cell proliferative responses. T-cell numbers decline with age in all children, but the decline in patients who have chromosome 22q11.2 deletion syndrome seems to be slower. In fact, adults who have chromosome 22q11.2 deletion syndrome have normal T-cell numbers for the most part. The slower age-determined decline in T-cell numbers in patients compared with controls is caused by the homeostatic proliferation of existing T cells[28]. Conclusion Association between CHDs and chromosome 22q11.2 microdeletions suggests that FISH analysis should be performed in CHD patients even when there are mild, unspecific extra cardiac anomalies. Due to the wide range and extremely variable expression of extra cardiac anomalies, a careful general clinical examination and gentle work up, together with genetic counseling in patients with or without syndromic CHD finding are compulsory. In cases with chromosome 22q11.2 microdeletion, both parents must be studied and an appropriate genetic counseling must also be offered to provide more accurate estimation of recurrence and possibility of prenatal diagnosis. Acknowledgment This work was partly funded by Tehran University of Medical Sciences (Grant no 1044). We would like to thank Mrs. Moqadam and Mrs. Hamidi for their help. We would also like to thank the scientists and staff of the Gene Clinic, Tehran, Iran, for their involvement in the FISH studies. References
© 2009 by Center of Excellence for Pediatrics, Children’s Medical Center, Tehran University of Medical Sciences,All rights reserved. The following images related to this document are available:Photo images[pe09002t1.jpg] [pe09002t2.jpg] [pe09002t3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}