 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
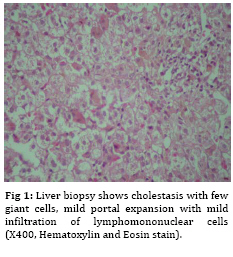
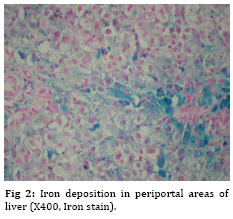
Iranian Journal of Pediatrics, Vol. 19, No. 2, June, 2009, pp. 193-195 Neonatal Giant Cell Hepatitis in an Infant with Cystic Fibrosis Gholam-Hossein Fallahi* 1,2, MD; Fatemeh Bazvand2, MD; Kambiz Eftekhari1,2, MD; Faezeh Ahmadi2, MD; Maedeh Ahmadi2, MD;Nima Rezaei2,3, MD 1. Department of Pediatric, Tehran University of Medical Sciences, Tehran, IR Iran Received: Aug 12, 2008; Final Revision: Dec 12, 2008; Accepted: Jan 20, 2009 Code Number: pe09032 Abstract Background: Cystic fibrosis is a hereditary disease of mucus and sweat glands characterized by respiratory infections and pancreatic insufficiency. Key Words: Cystic fibrosis; Cholestasis; Giant cell hepatitis; Neonatal hepatitis Introduction Cystic fibrosis (CF) is a fatal genetic disease of sweat glands and mucus secretion[1]. The abnormal mucus causes a variety of clinical manifestations, including recurrent respiratory infections, obstruction of the pancreas duct, disorder of bile secretion, and gastrointestinal obstruction. CF can present with different clinical features and severity from patient to patient [1]. While respiratory and pancreatic diseases are present in more than 80% of CF patients, liver involvement is less frequent, which is estimated to occur in only one third of the patients [2]. Liver diseases usually occur in adolescence, but are rare in infancy [3-5], which is one of the most important concerns in CF disease. In this report, we present an infant with CF who suffered from neonatal cholestasis associated with giant cell neonatal hepatitis. Case presentation An 1.5-month old girl, the first child of unrelated parents, was referred to our center with icterus and edema. She was well until the age of 1 month when she had acholic stools. After 10 days she showed icterus and was referred to our referral center in Tehran, Children’s Medical Center. She was hospitalized for further evaluation.In physical examination she had mild edema on limbs without hepatospenomegaly. Bilirubin assay revealed a high level of direct bilirubin (total bilirubin 20 mg/dl and direct bilirubin 15 mg/dl). During hospitalization the level of hemoglobin (Hb) dropped from 11 to 6 mg/dl, while the Coombs test was negative. Several blood transfusions were needed to raise Hb level to 7.5 mg/dl. Alanine aminotransferase (ALT) and aspartate aminotransferase (AST), γ-glutamyl transpeptidase (γGT)and alkaline phosphatase (Alk-Ph) were 26, 193, 88 and 603 IU/L, respectively. Total protein was 4 and albumin 2.09 g/dl. Liver, spleen, gall bladder and biliary ducts were normal in abdominal ultrasonography. Fat droplets were seen in stool examination. Chromatography of serum amino acids was normal and trypsin enzyme activity was in normal range. All other laboratory findings were also normal. The hepatobiliary iminodiacetic acid (HIDA) scanshowed grad І hepatocyte dysfunction with no discrete evidence of excretion into the bowel up to 24 hours. Because of edema, hypoalbuminia and anemia, CF was suspected, which was confirmed by two positive sweat tests. Liver biopsy (Figure 1 and 2) showed giant cell neonatal hepatitis with periportal iron deposition, maybe due to repeated transfusions. She was treated with fat soluble vitamins, zinc sulfate, creon and cimetidin. The bilirubin decreased (total bilirubin 9.3 mg/dl and direct bilirubin 7 mg/dl), while Hb increased to 8.9 mg/dl. She was discharged from hospital in good condition. Ursodeoxycholic acid was prescribed to improve bile secretion. Discussion Liver involvement in children with CF can be manifested in different features. While it may progress silently and only presenting as end-stage liver disease and portal hypertension[6], hyperbilirubinemia is the most common presentation of liver disorder in infancy[4]. Nevertheless, neonatal cholestasis is a rare condition[3,7]. Neonatal cholestasis occurs in the first three months of life, and biliary atresia and neonatal hepatitis are the most common causes of the disease[8], but CF is a rare underlying cause of this condition. Intra- and extra-hepatic impairment of biliary drainage is important in the pathogenesis of liver disease in cystic fibrosis[9]. Although several studies present a number of risk factors for developing liver disease in CF[4,10], our patient had none of the known factors. Neonatal hepatitis is a rare form of liver involvement in CF [4]. In the series study by Shapira et al, 5 of 12 patients with cholestasis had giant cell transformation[4]. The cholestasis may lead to liver failure even at the age of 6 weeks[3], while patient with infantile liver disease in CF seems to have good prognosis[4]. Abdominal ultrasonography, hepato-biliary scintigraphy and percutaneous liver biopsy are methods which could be useful in assessing cholestasis[8]. Because of bile secretion disorder in CF, the result of hepatobiliary scintigraphy may resemble biliary atresia and this has occurred in our patient. The diagnosis of giant cell neonatal hepatitis was confirmed in our patient after exclusion of other causes of cholestasis. Conclusion Although neonatal hepatitis is a common cause of neonatal cholestasis, it is quite rare in cystic fibrosis, which easily could be misdiagnosed. Early diagnosis and appropriate treatment could prevent further complications. Acknowledgment The authors would like to acknowledge Dr. Fatemeh Mahjoub for providing the histopathologic report of the patient. References
The following images related to this document are available:Photo images[pe09032f1.jpg] [pe09032f2.jpg] |
| |||||||||

{kind=link}
{kind=link}