 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Iranian Journal of Pediatrics, Vol. 20, No. 1, January-March, 2011, pp. 51-57 Detecting Common CFTR Mutations by Reverse Dot Blot Hybridization Method in Cystic Fibrosis; First Report from Northern Iran Mohammad-Reza Esmaeili Dooki1 ,MD; Haleh Akhavan-Niaki2, PhD, and Ali Ghabeli Juibary2, MD
* Corresponding Author; Address: Cellular and Molecular Biology Research Center, Babol University of Medical Sciences, Babol, Iran E-mail. halehakhavan@yahoo.com Received: Jun 01, 2010; Final Revision: Oct 29, 2010; Accepted: Nov 13, 2010 Code Number: pe11009 Abstract Objective: Cystic
fibrosis and its distribution vary widely in different countries and/or
ethnic groups. Common CFTR mutations were reported from Iran, but the
northern population was not or underrepresented in those studies. The
aim of this study was to determine the frequency of common CFTR mutations
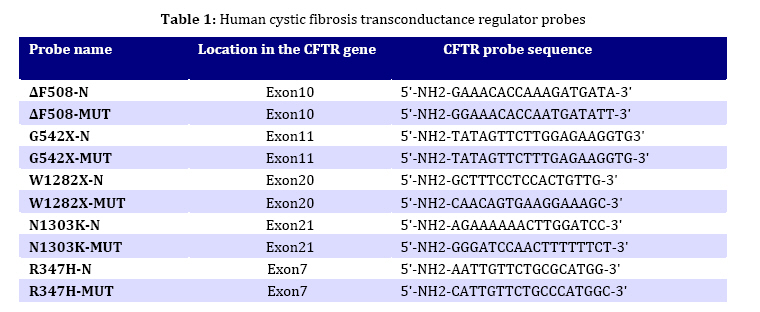
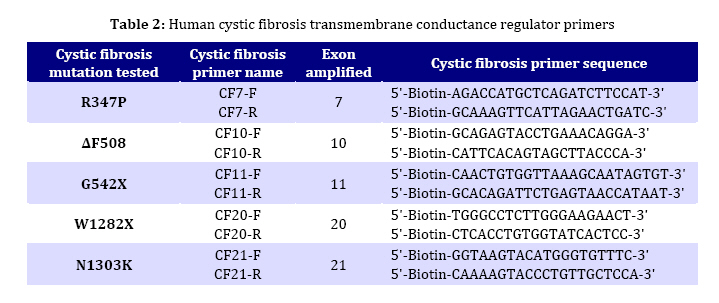
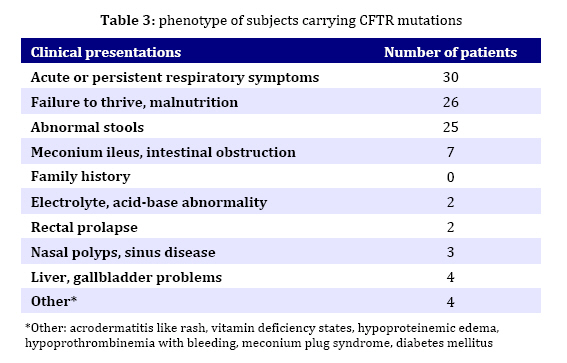
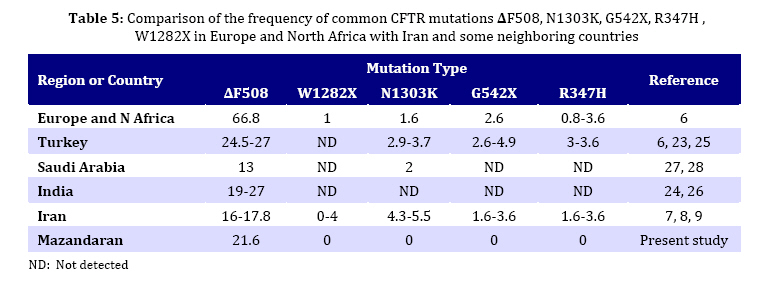
in children from northern Iran. Key Words: Cystic Fibrosis; CFTR; Genotype; DeltaF508-CFTR; Children; Iran Introduction Cystic fibrosis (CF) is an autosomal recessive disease caused by defect in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. The protein product of the gene is a chloride channel and a member of the ATP-binding cassette membrane transporter superfamily. The CFTR channel is critical for the normal function of epithelial cells in the lungs, pancreas, intestine, gall bladder, and sweat glands. Excess mucus in the respiratory system of CF patients facilitates chronic bacterial infections, leading to respiratory failure, which is the major cause of mortality. Most of the patients also fail to produce digestive enzymes in the pancreas, resulting in pancreatic insufficiency. Other clinical features are variably associated with the disease[1]. Approximately one in 2000 to 3000 newborns in populations of European ancestry are affected, and the average carrier frequency is about 1:25[2,3]. The p.F508del (∆F508) allele of the gene is the most common mutation observed worldwide and is probably very old, dating to pre-Neolithic times[2,4]. In addition to the ∆F508 mutation, more than 1000 other mutations in the CFTR gene have been identified (CF Genetic Analysis Consortium http://www.genet.sickkids. on.ca/cftr). These mutations vary greatly in their frequency and distribution, but most are very rare. Only four (p.G542X, p.N1303K, p.G551D and p.W1282X) have overall frequencies higher than 1%[5]. Intriguingly, p.G542X and p.N1303K are found on the same haplotype background as ∆F508, suggesting that they arose in the same population[6]. Few previous reports of CFTR mutations in Iran have been published[7-9]. In the present study, 5 of the 27 CFTR exons of 30 unrelated northern Iranian CF patients were analyzed with the aim of detecting common CFTR mutations in this population. In addition, the clinical presentations and laboratory findings were studied based on hospital and outpatient records. We selected five mutations, deltaF508, N1303K, G542X, R347H and W1282X based on previous reports in Iran and neighboring countries. The knowledge of the mutation spectrum and its frequency in this population will improve the diagnosis and medical care of CF patients and could also be used as a basis for carrier screening and prenatal diagnosis programming in Iran. Several techniques such as allele specific oligonucleotide (ASO) dot-blot, reverse dot-blot, amplification refractory mutation (ARMS), and an oligo-ligation assay, are available to detect the most common mutations[10]. The reverse dot-blot (RDB) technique is one of the most widely used techniques to diagnose CF[11-14]. This technique was evaluated as routine first-line analyses of the CFTR gene status[12,15]. RDB is more advantageous than other techniques since it does not necessitate the use of radiolabled probes as in Allele Specific Oligonucleotide assay and the use of positive controls to show the proper functioning of PCR reaction in each tube as in ARMS assays, since in ARMS technique, results are based on the absence of PCR products[13]. In this study all 5 mutations were screened simultaneously using reverse dot blot (RDB) assay. Subjects and Methods Patients: The 30 patients were diagnosed as cystic fibrosis based on elevated sweat chloride values (>60 mEq/L) and were 6 weeks to 11 years old. Many patients had pulmonary complications and pancreatic insufficiency. The patients were recruited from a pediatric hospital (Amirkola Children's Hospital) in Mazandaran province, northern Iran. Some of them were diagnosed by the pediatric gastroenterologist in office. The patients originated from many different regions of Mazandaran. Families of the patients were informed of the nature of the research and consented to participate in this study. Molecular Analysis: DNA from the leukocytes of peripheral blood was prepared by the salting out protocol. Mutation detection was performed using Reverse Dot Blot (RDB) procedure, as described by Lappin et al[16]. In this assay, oligonucleotide probes were synthesized using a C6-amino-linker on the 5' end of the product and were attached onto Biodyne C nylon membrane (Pall Corporation) activated by 1-Ethyl 3-Dimethyl Aminopropyl Carbodiimide (EDAC). The target DNA was amplified with 5' biotinylated primers and hybridized to immobilized oligonucleotides on the membrane. Hybridization was detected by adding streptavidin-horseradish peroxidase to the membrane where it bound to biotinylated DNA that was hybridized to the oligonucleotides. A positive signal could then be obtained by a simple non-radioactive colorimetric reaction. CFTR amplifications took place in 50-µL reactions containing 1X PCR buffer (10 mmol/L KCl, 10 mmol/L Tris-HCl, pH 8.3, 1.5 mmol/L MgCl2) and 6 mmol/L MgCl2, 200 µmol/L each of dNTPs, 10 pmol of each primer, and 1.5 units of Taq DNA polymerase. All PCR reagents (except primers which were from MWG, Germany) were from Roche Company. PCR was performed in a Techgene thermocycler from Techne Co., UK). Thermo-cycling conditions were uniform for all reactions and included an initial denaturation step at 95°C for 3 minutes, followed by 38 cycles of denaturation at 95°C for 30 seconds, annealing at 54°C for 30 seconds and polymerization at 72°C for 30 seconds. Cycling culminated with a final extension at 72°C for 4 minutes. PCR products were visualized after electrophoresis on 1.5% Agarose LE gel under 302nm UV lamp. The hybridization reaction took place after adding the pooled PCR products to Biodyne membranes pre-wet with hybridization buffer [2X saline sodium citrate (SSC), 0.1% sodium dodecyl sulfate (SDS)] at 42°C, The amplicons and membranes were boiled and permitted to hybridize for 1 hour at 42°C. Hybridization solutions were poured off and membranes were washed twice in excess of wash buffer (0.75_ SSC, 0.1% SDS). Membranes were then agitated in a freshly prepared conjugate solution (1:4000 dilution of streptavidin-horseradish peroxidase conjugate, (Boehringer Mannheim, Part No. 1089153), in 0.5X SSC, 0.1% SDS for 30 minutes at room temperature. At the end of the incubation, membranes were washed three times for 3 minutes each with 0.5X SSC, 0.1% SDS; then twice for three minutes each with 0.1 mol/L sodium citrate, pH 5.0. The membranes were then exposed to 0.01% w/v tetramethylbenzidine dihydro-chloride (TMB) substrate (Sigma T-8768) in a freshly prepared, dilute solution of hydrogen peroxide (0.00225% v/v) in 0.1M sodium citrate, pH 5.0 with agitation for 10 to 20 minutes. Color development was stopped by rinsing the membranes with distillated water. The details of probes and primers (MWG Biotech, Germany) used in this study are presented in Tables 1 and 2 respectively and were described previously by Lappin et al[16]. Findings Phenotype Analysis: 30 unrelated patients (17 males and 13 females) aged 6 weeks to 11 years originating from Mazandaran province, Iran were analyzed in this study. Half of the patients were issued from consanguineous marriage (mostly between first cousins). All patients showed acute or persistent respiratory symptoms. 26 patients had failure to thrive and malnutrition and 25 patients had abnormal stools. None of the patients had a previous family history of CF. Other manifestations such as meconium ileus, intestinal obstruction, electrolyte abnormality, rectal prolapse, nasal polyps, sinus disease, diabetes mellitus, liver or gallbladder problems were observed in a small number of patients. The phenotype of subjects carrying CFTR mutations is reported in Table 3. Genotype Analysis: Mutation screening of the CFTR gene in 60 alleles by reverse dot blot hybridization for five common mutations showed that 13 (21.6%) alleles were ∆F508. Six patients were homozygous for ∆F508 and one patient was compound heterozygous for whom only one allele was specified. The other four mutations tested (N1303K, G542X, R 347H and W1282X) were not encountered in these patients. Genotype-Phenotype Correlation: Table 4 shows phenotypes of 7 patients carrying deltaF508 mutation. All had respiratory difficulties and failure to thrive. Six of them evacuated abnormal stools and 4 had meconium ileus. Other manifestations such as edema and hypoproteinemia, liver and gallbladder problems, rectal prolapse, or diabetes mellitus were encountered in a lower incidence (one or two patients). Discussion Few molecular studies are performed in Iranian CF patients[7-9]. As Mazandaran CF patients were underrepresented (2 out of 69 patients)[9] or not included in those studies, and regarding the high genetic heterogeneity observed among different provinces of Iran in other disease causing genes[17- 19] and also considerable heterogeneity in the nature of CFTR mutations observed among Iranian cystic fibrosis patients[7-9], the molecular characterization of CF in Mazandaran province seems a necessity. Using Reverse Dot Blot method, 5 common CFTR mutations were analyzed in this study in 30 CF patients (13 males and 17 females) aged 6 weeks to 11 years, originating from Mazandaran province and presenting acute or persistent respiratory symptoms and failure to thrive. This method is a simple, rapid and reliable method which allows simultaneous detection of up to 20 different mutations in a single hybridization assay[13]. Delta F508 mutation was present in 13 (21.6%) alleles tested by RDB assay. Six patients were homozygous and one was compound heterozygote for this mutation. The other four mutations tested: N1303K, G542X, R347H and W1282X, were not found in these patients. These mutations were previously reported in most parts of the world[21,22] including neighboring countries[23-28] and also other provinces of Iran[7-9]. Only one disease-causing mutation was identified in this study. Although the frequency of the ∆F508 among Mazandarani patients was lower (21.6%) than its frequency in European countries, it was still the most frequent mutation among those reported to date in Iran. Previous studies representing overall Iranian CF patients reported a frequency of 16-17.8% for this mutation. This difference could be due to the small number of Mazandarani patients in this study and suggests heterogeneity of Iranian provinces regarding CF mutations. Moreover, its frequency is comparable with those reported in several countries in west Asia, north Africa and Indian subcontinent (Algeria 16.7%, Tunisia 18%, Turkey 24-27%, Saudi Arabia 13%, Pakistan 17%, India 19-27%[6,20,21, 24,26-30]). Previous studies showed that the mutation spectrum in Iran had a significant overlap with that observed in Turkey and was less similar to its spectrum observed in Europe. Specifically, the four most common Turkish mutations were found in Iran, including ∆F508, c.1677delTA, p.G542X, and c.2183AA>G. The "Mediterranean mutation", p.G542X, is reported to be of Phoenician origin[7-9,29,31]. Table 5 shows the comparison of the frequency of all five mutations considered in this study in Iran and some neighboring countries. Regarding the sample size in this study, underestimation of the other four mutations analyzed may be possible due to their low frequency. All patients presented acute or persistent respiratory symptoms and more than 80% had failure to thrive, malnutrition and abnormal stools. Only 23.3% of patients (7 cases) had meconium ileus and intestinal obstruction among which 4 had homozygote ∆F508 genotype. The high prevalence of meconium ileus among ∆F508 patients was also reported in other studies[32-33]. Liver disorders which are present in 2-3% of children and 5% of adults occur in 25% of CF patients[34]. In this study 4 children presented liver disorders (13.3%) among which 2 had ∆F508 homozygote genotype. Frequency of carriers of a disease-causing CFTR mutation in Iranians (1:40)[8] is very similar to that in European populations (1:25). Consequently, it is believed that the incidence of cystic fibrosis is also similarly high, and that the low incidence commonly believed to be associated cystic fibrosis is also similarly high, and that the low incidence commonly believed to be associated with this non-European population is likely to be due to under-diagnosis. Moreover, intermediate sweat chloride levels may exist in some CF patients[35], leading to misdiagnosis of these patients if the selection criteria is based on sweat chloride positive test as is the case in this study. Furthermore, the frequency of CF may be higher in some isolated populations due to consanguinity, as has been previously reported in an isolated population[36]. The same assessment has been proposed for populations of Turkey and Saudi Arabia[27-29]. Therefore, in addition to Europe and the United States, CF is likely to be prevalent in many other countries as well, although the mutation spectrums may be different. Thus ascertainment of CFTR mutation carrier frequencies and CF incidence among Iranians seems to be a necessity. Conclusion Our mutation detection efficiency was rather low (21.6%). However, we cannot assure how much clinical misdiagnosis contributed to this. ∆F508 was the only mutation detected and showed a frequency comparable to that reported in overall Iranian population and neighboring countries. Identification of the remaining mutations and their frequency requires to design appropriate tests to improve the clinical diagnosis and establishing CF prevention programs by carrier screening and prenatal diagnosis. Acknowledgment We thank Mr. Ali Banihashemi, Mrs. Mandana Azizi and Mrs. Beheshteh Asghari for their technical assistance. This study was authorized by the Ethic Committee of Babol University of Medical Sciences. Conflict of Interest: None References
Copyright 2011 - Iran Journal of Pediatrics The following images related to this document are available:Photo images[pe11009t2.jpg] [pe11009t1.jpg] [pe11009t4.jpg] [pe11009t5.jpg] [pe11009t3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}