 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Iranian Journal of Pediatrics, Vol. 20, No. 1, January-March, 2011, pp. 77-82 Assessment of Thyroid Function in Children Aged 1-13 Years with Beta-Thalassemia Major Ayfer Gözü Pirinççioğlu1, MD; Turgay Deniz1, MD; Deniz Gökalp2, MD; Nurcan Beyazıt1, MD; Kenan Haspolat1, MD, and Murat Söker1, MD
* Corresponding Author; Address: Department of Pediatrics, Faculty of Medicine, University of Dicle, 21280, Diyarbakir, Turkey E-mail: ayfergozu@hotmail.com Received: Mar 17, 2010; Final Revision: Oct 18, 2010; Accepted: Nov 02, 2010 Code Number: pe11013 Abstract Objective: Hypothyroidism
usually appears in the second decade of life and is thought to be associated
with iron overload in patients with thalassemia major. This study aimed
to evaluate thyroid dysfunctions in patients with beta-thalassemia
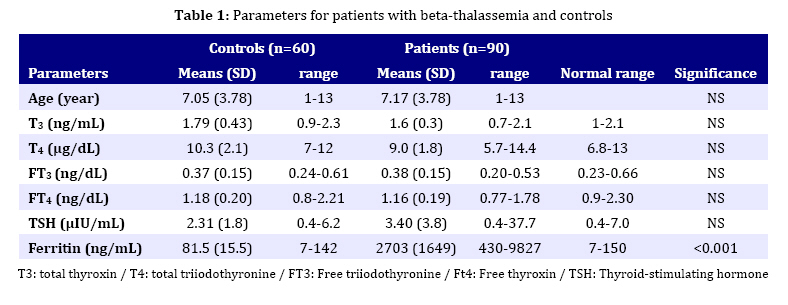
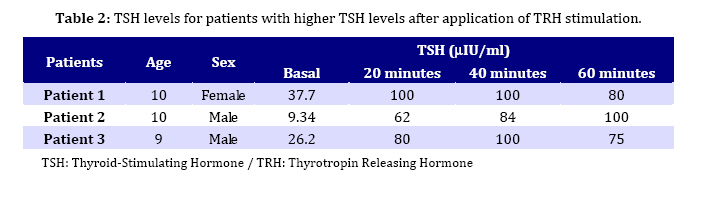
major and to see if they appear in the earlier period of life. Key Words: Beta-Thalassemia; Hypothyroidism; Iron Overload; Chelation Therapy; Spelenectomy Introduction Beta-thalassemia major is an autosomal recessive hereditary anemia, which is incurable, caused by defective synthesis of hemoglobin, ineffective erytropoiesis and rapid erythrocyte breakdown, resulting in advanced heart failure and death in early childhood[1,2]. The combination of transfusion and chelation therapy has dramatically extended the life expectancy of thalassemic patients who can now survive into their fourth and fifth decades of life[3]. Nevertheless, frequent blood transfusion in turn can result in iron overload which may lead to various complications, including various endocrine complications such as thyroid dysfunction[4]. So, bone marrow transplantation remains the only definitive cure for thalassemics at present[5-7]. However, this application involves its own fair share of complications[8]. The widespread form of thyroid dysfunction seen in thalassaemics is primary hypothyroidism-caused abnormalities of the thyroid gland which results in insufficient production of the thyroid hormones. Nonetheless, the frequency of hypothyroidism shows a discrepancy depending on the region, quality of management and treatment protocols[9,10]. Thyroid dysfunctions are well documented in patients with thalassemia major requiring frequent and recurrent blood transfusions. These have recently been discussed in details in the literature[11-18]. It is a general belief that thyroid dysfunctions appear with a frequency of 13-60% in these patients after 10 years of age regardless of difference in the rate of prevalence, largely as in the form of subclinical hypothyroidism[9,10]. This work aimed to investigate the frequency of hypothyroidism in children with beta-thalassemia major in their earlier period. Subjects and Methods The parent of all patients gave informed consent prior to the study entry and the study was conducted in accordance with the Declaration of Helsinki. Ninety patients (55 boys and 35 girls) with beta-thalassemia major aged 1-13 years (mean age 7.17±3.78 years) were enrolled in this study, admitted to the Department of Pediatrics, Faculty of Medicine University of Dicle between 21/03/05 and 31/07/09. An age-sex matched control group of 60 healthy children (36 boys and 24 girls with a mean age of 6.98±3.66 years) was also formed. Patients with beta-thalassemia minor and intermedia and those with acute illness, any hormonal therapy or with a family history of hypothyroidism were excluded from the study. Diagnosis of beta-thalassemia major was based on family history, complete transfusion dependence and hemoglobin electrophoresis. Blood transfusion was applied as packed treated red cells. Patients received an average of 7.6±2.4 mL packed cells per kilogram body weight with a mean of 17.2±5.4 transfusions over a period of 12 months to maintain hemoglobin concentration not to exceed 9.5 g/dL. After transfusion, chelation therapy was employed using deferoxamine by subcutaneous infusion with a dose of 20-40 mg/kg/24 hr over 8-12 hr via portable infusion device. Thyroid function and iron load status were evaluated by measurements of serum total thyroxin (T3), serum total triiodothyronine (T4), serum free triiodothyronine (FT3), serum free thyroxin (FT4), thyroid-stimulating hormone (TSH) and serum ferritin levels. A standard TRH testing (7μg/kg bolus intravenously) was applied to three patients with high levels of TSH. Serum TSH levels were measured at 0, 20, 40 and 60 minutes intervals after TRH administration. Blood samples were taken from all patients at least 15 days after the transfusion, preferably after an overnight fasting on the transfusion day. Total serum T3, T4, FT3, FT4, TSH and ferritin levels were studied in basal blood sample employing Electrochemiluminescence Immuno-assay (ECLIA) method using Roche Diagnostic E 170 Autoanalyzer. Antithyroglobulin and anti thriod peroxidase (TPO) antibody titers were measured by radioimmunoassay (RIA). The thyrotropin releasing hormone (TRH) warning test was applied to the patients whose basal TSH value was detected to be over the age-appropriate reference range. Statistical analyses were carried out using the software packages SPSS 12.0 (SPSS, Inc., Chicago, USA) using Student’s t-test and Spearman correlation and regression analyses were applied in statistical evaluations. Statistical significance was accepted as P<0.05. Results are expressed as mean±standard deviation (SD). Findings All patients underwent comprehensive physical examination and were examined if they had any other complaints except beta-thalassemia at the time of presentation to the Hematology Polyclinic. The result of physical examination showed that none of the patients have had goiter. Antithyroglobulin and anti-thyroid peroxidase antibody titers were negative in all patients. A total of 90 patients were enrolled in the study, 35 (38.9 %) of them were female and 55 (61.1%) male. Mean age of females and males was 7.16±4.06 and 7.61±4.26 years respectively. Mean hemoglobin (Hb) levels before and after blood transfusions were reported as 8.7±0.6 and 12.8±1.2. This value was 13.3±1.4 in the control group. Biochemical parameters measured in serum of patients and healthy children are recorded in Table 1. Serum ferritin levels were found as 0-1000ng/mL in 6, 1000-2000 ng/mL in 22, 200-4000 ng/mL in 35 and >4000ng/mL in 12 patients. The TSH levels were higher than normal range only in three patients. Low TSH levels were not detected. The total T3, T4, serum FT3 and FT4 levels were normal in these 3 patients. The TRH stimulation test was applied to them. Data are presented in Table 2. All three patients were classified as subclinical primer hypothyroidism, normal FT3 and FT4, increased basal TSH levels and increased TSH response to TRH. They were started on treatment with Levotrion. The correlation between parameters studied was as follows. A negative correlation between T3 and the age of the patients (r=-242; P=0.037), and between TSH and T4 (r=-0.268; P=0.002) was observed while a positive correlation between T3 and FT3 (P<0.001), between T3 and FT4 (r= 0.241; P=0.035), between FT3 and FT4 (r=0.556; P<0.001), between ferritin and FT3 (r=0.261; P=0.024), ferritin and the age of patients (r=0.309; P=0.007) was observed. It was observed that transfusion frequency had a significant increasing effect on the serum ferritin level (P<0.001) as well as on basal TSH levels (P=0.037). Splenectomy was applied to 20 patients (22.2%) with an average age of 10.60±4.10 years. The effect of blood transfusion frequency (BTF) on laboratory parameters was also investigated. It was found that BTF only influences ferritin levels. Those receiving blood transfusion every 3-4 weeks had higher ferritin levels (3745±1926 ng/mL) compared with those (2008±946 ng/mL) receiving transfusion every 4-12 weeks (P=0.05). It was also seen that there was a correlation between the application of splenectomy and blood transfusion rate (P=0.05). 16 out of 20 patients treated with splenectomy received blood transfusion every 4-12 weeks while 4 out of 20 received blood transfusion every 3-4 weeks. The effects of chelation therapy using desferrioxamine (DFO) and supplementary therapy (zinc, folic acid, vitamin C, vitamin E) on parameters and their relationship with other applications such as splenectomy and BTF were studied and analyzed. 55 patients (61.1%) were receiving DFO while 35 (28.9 %) patients were not on use of DFO. It was found that the use of DFO had no statistically significant effect (P>0.05) on the biochemical parameters but a significant correlation (P=0.001) was found between the age of patients and DFO treatment. The mean age of those treated and not treated with DFO was 8.68±3.62 and 4.76±2.61, respectively. It was indicated that supplementary therapy has not a significant effect on biochemical parameters and the applications (P>0.05). It was also observed that there is no effect of the sex of patients on biochemical parameters and applications such as splenectomy, BTF, DFO treatment and supplementary therapy. Discussion One important aspect of management in polytransfused thalassemic patients is early recognition and treatment of endocrine dysfunction. This is particularly true for thyroid dysfunction, because hypothyroidism could be associated with growth problems so commonly seen in these patients. The present study showed that T3, T4 and FT3, FT4 levels in all patients were in normal range compared with those in controls while TSH level was found to be high in only 3 patients and normal in the remaining 87 patients. However, mean TSH and ferritin levels were significantly higher in patients compared to those of controls (Table 1). Since anti-thyroglobulin and anti-thriod peroxidase (anti-TPO) antibody titers were negative in three patients with high TSH they might be classified as subclinical primer hypothyroidism. Other manifestations of hypothyroidism such as overt primary hypothyroidism and secondary hypothyroidism were not found in the presented patients. In a recent paper, De Sanctis et al[3] and Malik et al[10] reported the ratio of primary hypothyroidism as 2.1% (in a study group of 238 patients aged 2-17 years) and as 25.7% (in a study group of 70 patients aged 5-14 years) with TM. In our study, this was quite low but it is still comparable with the work of De Sanctis. The reason for the lower frequency may be attributed to the fact that the majority (80%) of patients in the present work were under 10 years old. Hypothyroidism may be partly related to the accumulation of iron in thyroid glands due to blood transfusion by iron overload leading to gland dysfunction[19]. Iron overload of tissue is the most important complication of beta-thalassemia and is a major subject of management[20]. Although most clinical signs of iron loading do not appear until the second decade of life in patients with inadequate chelation, evidence from serial liver biopsies in very young patients presents that the toxic effects of iron begins much earlier. After approximately one year of transfusions, iron starts to be accumulated in parenchymal tissues, where it may bring about substantial toxicity as compared with that within reticuloendothelial cells[21,22]. Despite the reports relating endocrine dysfunction with iron overload it was recently demonstrated that the degree of iron overload, at least reflected by ferritin levels, was not associated with the development of endocrine complications[23,24]. Our study also showed that there is no correlation between serum ferritin level and thyroid function but a positive correlation between transfusion frequency, an indirect indication of ferritin overload, and hypothyroidism. It indicated that TSH levels were higher in the patients receiving blood transfusion every 3-4 weeks compared to those receiving blood transfusion every 4-12 weeks. The absence of the relationship between ferritin and hypothyroidism may be explained by suggesting that the damage of endocrine glands caused by chronic hypoxia is more pronounced than that caused by hemosiderosis as a consequence of the collapse of iron. Thyroid failure is expected to be more prevalent in older patients (as it is true for other endocrine deficits) and hence a correlation between older age and reduction of serum T3 and FT3 levels is predicted. The mean age of patients in our work (7.17 years old) is lower than in most studies. Since a large number (80%) of patients in our study were under the age of 10 years (the age of all 3 patients with thyroid dysfunction was around 10 years), the relationship between the age of patients and thyroid function may not be observed. Gulati et al also mentioned the possibility of endocrine dysfunction in patients with thalassemia in earlier periods[4]. Consistently the study illustrated that thyroid functions are not correlated with the age of patients. However, it was found that ferritin level is correlated with the age. This is inevitable since the older patients would have longer blood transfusion period and as a result chelation therapy is recommended to reduce iron overload. Although the use of iron chelation therapy has been known to delay or reverse the development of iron-induced cardiac damage and to improve survival[25], the ability of deferoxamine (DFO) to prevent iron-induced endocrine complications is less well defined, but with increased survival, the consequences of endocrine failure become more important. In our study, three patients assigned as subclinical primary hypothyroidism were approximately 10 years old and they were not receiving regular DFO treatment. The study also showed that DFO has no effect on any parameter studied. We also detected that there was no significant relationship between the gender and thyroid dysfunction. However, higher incidence of thyroid dysfunction was reported in females[26]. It was shown that splenectomised patients (n=20) were older (10.6±4.1) than those (6.6±3.38) not-splenectomised (P<0.001) and also found that splenectomised patients had higher blood transfusion rate (P=0.05). The result also indicated that splenectomy has no effect on thyroid function, which is consistent with previous studies[27]. Further investigation is needed to associate the relationship between iron-overload and thyroid function in children with beta-thalassemia. Since serum ferritin is a poor marker in heavily overloaded individuals, it is possible that the effect of iron on thyroid functions was underestimated in this study. Conclusion Thyroid dysfunction in thalassemia may start early in life though with a low frequency. Therefore, thyroid function should be followed periodically, particularly when other iron overload-associated complications occur. Early recognition and hence prevention of these complications might help improve the quality of life of these patients. Acknowledgment The authors wish to thank all the thalassemic patients and controls that took part in this study and the Department of Pediatrics, Faculty of Medicine, University of Dicle for giving permission to access data and to carry out this work and also for their help and guidance. Conflict of Interest: None References
Copyright 2011 - Iran Journal of Pediatrics The following images related to this document are available:Photo images[pe11013t2.jpg] [pe11013t1.jpg] |
| |||||||||

{kind=link}
{kind=link}