 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Iranian Journal of Pediatrics, Vol. 20, No. 1, January-March, 2011, pp. 107-112 Acute Hemorrhagic Edema of Infancy; a Report of Five Iranian Infants and Review of the Literature Mohammad-Hassan Moradinejad1,2, MD; Pegah Entezari2, MD; Fatemeh Mahjoub3, MD, and Vahid Ziaee1,2, MD
Received: Feb 02, 2010; Final Revision: Aug 05, 2010; Accepted: Sep 17, 2010 Code Number: pe11019 Abstract Background: Acute
hemorrhagic edema of infancy (AHEI) is a benign self limiting leukocytoclastic
vasculitis in young children. Serious complications, e.g. renal and
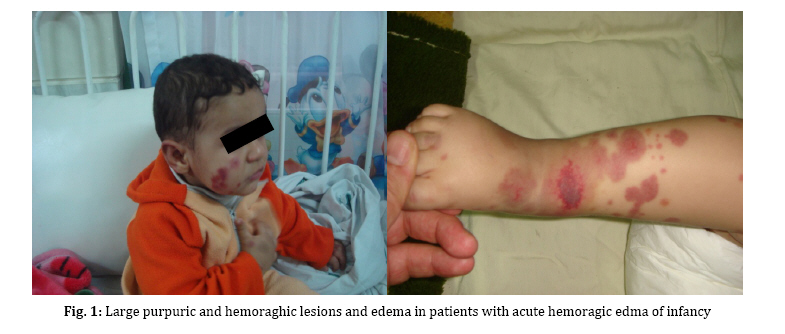
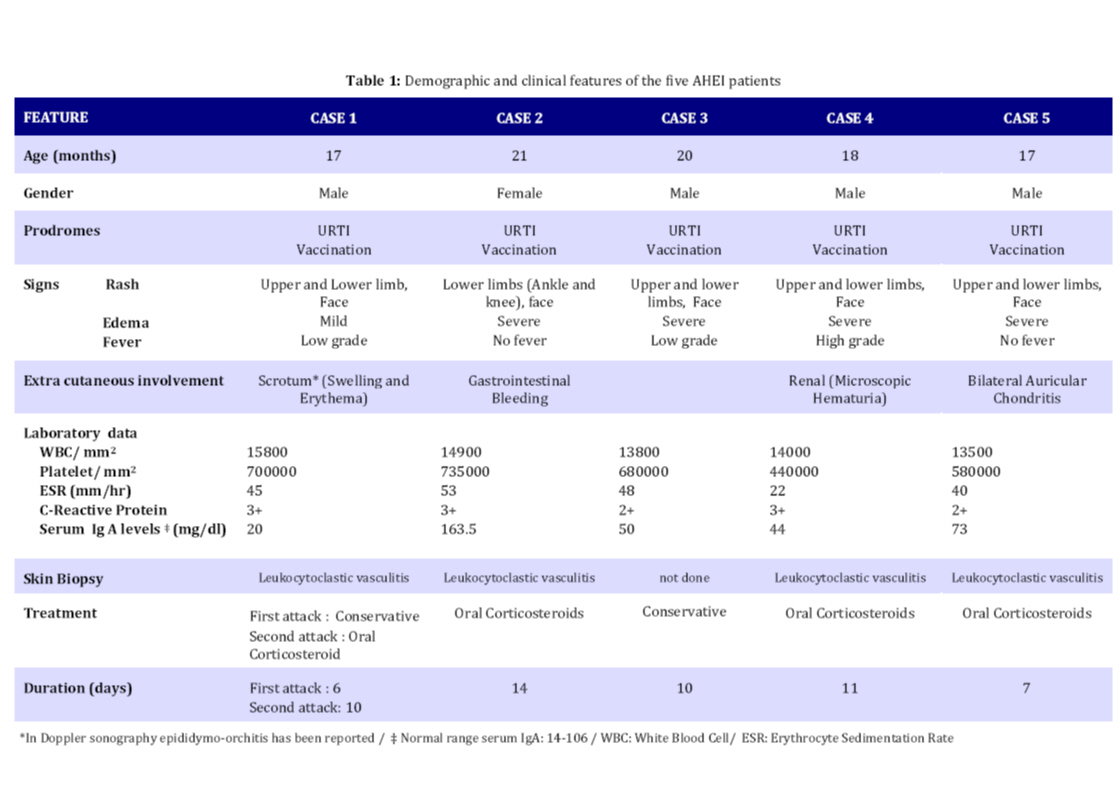
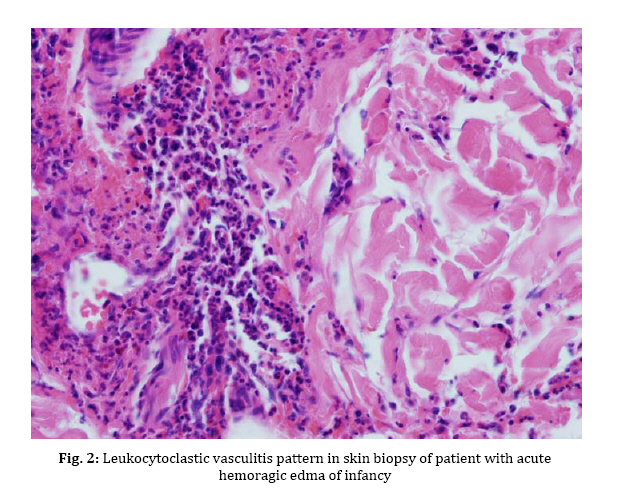
gastrointestinal involvement, are not usually detected in AHEI patients. Key Words: Hemorrhagic Edema; Henoch-Schoenlein Purpura; Leukocytoclastic Vasculitis; Gastrointestinal Bleeding; Hematuria Introduction Acute hemorrhagic edema of infancy (AHEI) is a dermal leukocytoclastic vasculitis with striking cutaneous lesions presenting in patients younger than two year old with an otherwise good general condition. Diagnosis of the syndrome is based on clinical symptoms and histological findings[1]. Purpuric palpable rash, edema and low grade fever are the typical clinical triad of AHEI syndrome. On the other hand, leukocytoclastic vasculitis (LCV), perivascular neutrophilic infiltration and in some cases fibrinoid necrosis are among the histological findings of AHEI. This syndrome has a good prognosis ending in complete cure[2]. Patients with AHEI usually don’t need to be treated with steroids or antihistamines, supportive wound care is efficacious[3]. Thus, it is important to correctly diagnose the syndrome and differentiate it from its important differential diagnosis, Henoch-Schoenlein Purpura (HSP) which is commonly seen in 3 to 6-year-old children, to avoid systemic treatment[3]. In this article we report five cases of AHEI referred to our hospital in one year; one of them was a typical case of AHEI, the second one had gastrointestinal bleeding and the third case experienced scrotal edema and recurrent attack. Fourth patient experienced bilateral auricular chondritis and the last one had microscopic hematuria. The medical treatment for each patient was different due to special indications. Case Presentation From January 2008 to January 2009, five cases with AHEI were referred to our hospital. They were 17 to 21 months old. Four (80%) were male and one (20%) female. All patients had edema and large purpuric lesions (Fig. 1). One patient had high grade fever (temperature >39°C) while two others had low grade fever (temperature <38.5°C). Despite dramatic appearance of the lesions, patients were otherwise well. The results of complete blood count, urine analysis, serum immunoglobulin levels and coagulation profile are shown in Table 1. Skin biopsy of four patients revealed leukocytoclastic vasculitis (Fig. 2). Skin biopsy was not done in one patient, since he presented typical features of AHEI and rapid regression of the lesions occurred without any medication. Four patients showed unusual symptoms: Case 1 experienced a recurrent attack with scrotal erythema and edema. Doppler ultra-sonography of the testicles showed epididymo-orchitis. Case 2 presented with blood-streaked stool at admission, Upper gastrointestinal studies and barium enema revealed no abnormal findings. Abdominal ultra-sonography was also normal. Occult blood test in stool was positive. No Coxsackie virus or Rotavirus was found in stool examination. Case 4 had renal involvement as microscopic hematuria and Case 5 had bilateral auricular chondritis. Patients underwent conservative treatment or systemic corticosteroid therapy according to severity of skin lesions, recurrence and visceral involvement. Skin rash and edema completely resolved in all cases within 6 to 14 days (median 10 days). On follow up no relapse occurred in four patients, Case1 experienced a second attack two weeks after the first one. Because of rapid progression of the lesions over limbs, face and scrotal area, systemic corticosteroids were administered. The symptoms started to resolve after 3 days. The demographic, clinical and laboratory features and the treatment chosen for each patient are depicted in Table 1. Discussion AHEI is a leukocytoclastic vasculitis defined for the first time by Snow in 1913[4]. Since then, remarkably few cases of AHEI have been reported, which could be the result of misdiagnosing the disease[5]. AHEI typically presents with a triad of edema, low grade fever and rosette-shaped skin lesions in a nontoxic 4 to 24-month-child[2]. Most of the time the symptoms last for a mean period of 1 to 3 weeks with spontaneous regression without sequel[2,5,6]. Skin lesions are usually targetoid, palpable rashes of 1 to 6 centimeters in diameter and are mostly detected on face and extremities[2]. Trunk, mucosal surfaces and viscera are typically spared, but some studies nevertheless report such involvement [6]. In this study, our patients presented with the typical triad. Two cases (Case 2 and 5) were afebrile and one (Case 4) had high grade fever at the time of admission, which is common in AHEI[2]. Our patients were 17 to 21 months old and had typical lesions of AHEI in typical parts of the body; visceral involvement was detected in two cases (Case 2 and 4). There was no trunk or mucosal involvement in our study. It took almost 10 days for the disease to cure completely. The etiology of AHEI has yet to be defined, but it is believed that infections [mostly upper respiratory tract infection (URTI) and urinary tract infection (UTI)], medications (especially antibiotics), and less commonly, immunization are factors that have a triggering role[3,7]. With clinical observation, some clinicians have deducted from the coinciding age of the children and their immunization schedule that immunization might be a trigger factor for AHEI[2]. In our study, all cases had URTI prior to skin lesions onset. All of them had immunization as scheduled. There was a history of antihistamines and acetaminophen (paracetamol) for URTI and phenobarbital for neonatal seizure in one case (Case 3). Among these medications, paracetamol (acetaminophen) is the only known trigger for AHEI[2]. Diagnosis of AHEI is based on clinical features of the syndrome and also on the histological changes of small vessels as leukocytoclastic vasculitis in dermis, i.e. perivascular neutrophilic infiltration with numerous scattered nuclear fragments at places invading the vascular wall and resulting in fibrinoid necrosis[8,9]. Laboratory data are often non diagnostic in AHEI; increased erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP), mild thrombocytosis and leukocytosis are the only abnormal findings that are usually reported in AHEI[2]. Serum complements, immunoglobulin (Ig) G, A and M are frequently within normal range or slightly elevated[6]. In patients with clinical suspicion of AHEI, complete blood count, urine analysis and basic coagulation profiles should be done to exclude other possible diagnoses[2]. In our study, mild leukocytosis, thrombocytosis as well as elevated ESR and CRP were detected in all cases. Mild elevation of serum IgA titer in Case 2 and microscopic hematuria in Case 4 was detected. Epididymo-orchitis was revealed in Doppler ultra-sonography of the testes in Case 1. Other laboratory data were within normal range in this study. Treatment of AHEI patients is based on conservative approach. No treatment with corticosteroids or antihistamines is needed and topical wound care is efficient most of the time[3]. Rapid progression of the syndrome could nevertheless be an indication for steroid therapy. As reported in one study, dramatic symptom regression occurred just 24 hours after starting systemic steroids[10]. Moreover, with some serious complications such as gastrointestinal bleeding, renal involvement or articular and cartilage involvement, prescribing medications (e.g. nonsteroidal anti-inflammatory drugs in articular involvement and systemic corticosteroids in gastrointestinal bleeding and hematuria) is necessary[2,11]. In our study, treatment was different according to the clinical presentation. We had conservative approach in one case. Systemic corticosteroids were given to other four patients due to visceral involvement, rapid progression of lesions in recurrent attack or severe cutaneous lesions. AHEI patients rarely experience recurrent attacks[2] and the syndrome typically resolves uneventfully, though there are few reports of serious complications in AHEI such as scarring lesions[12] and renal[13], gastrointestinal or cartilage involvement[11]. In this study we had relapsing attack and epididymo-orchitis (Case 1), gastrointestinal bleeding (Case 3), renal involvement (Case 4) and bilateral auricular chondritis (Case 5) which are not commonly seen in AHEI. There are several differential diagnoses for AHEI, HSP being the most important one[3]. Because of the similarities between the two syndromes, there has been a conflict between researchers about acknowledging AHEI as a separate entity or as a variant of HSP[14,15]. It is important to differentiate these syndromes from each other, since the medical approach to each one is quite different[2]. While AHEI does not require further treatment most of the time, HSP should be treated with systemic corticosteroids as soon as the syndrome is detected. HSP and AHEI are both recognized as rheumatologic diseases with leukocytoclastic vasculitis and skin lesions[2], but there are some features that may help differentiate HSP from AHEI. First of all, the former occurs more often in 3 to 6-year-old children, while the latter presents in younger , 3 to 24-month-old ones[16]. Furthermore, HSP presents with polymorphic lesions on the buttock and the extensor surfaces of the legs, with the face being usually spared[8]. Visceral, gastrointestinal and renal involve-ments are frequent complications of HSP[16]. On the other hand, AHEI has typically more monomorphic lesions on the extremities, the face and the scrotal area with more edema of the involved organs in comparison with HSP patients[8]. Extra-cutaneous involvement rarely occurs in AHEI[10,17]. A significant increase in IgA serum level is detectable in HSP, while the immunoglobulin serum levels are normal to slightly elevated in AHEI[10]. In contrast to AHEI, IgA vessel deposition is frequently seen in HSP, while there is typically no IgA deposition in AHEI[10]. The clues mentioned above led us to the diagnosis of AHEI rather than HSP in our patients. Conclusion It is important to correctly diagnose AHEI to avoid using unnecessary medications in this benign syndrome while closely monitoring them for serious complications. References
Copyright 2011 - Iran Journal of Pediatrics The following images related to this document are available:Photo images[pe11019f1.jpg] [pe11019f2.jpg] [pe11019t1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}