 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Iranian Journal of Pediatrics, Vol. 20, No. 1, January-March, 2011, pp. 121-125 Ectodermal Dysplasia Showing the Clinical Overlap between Hay Wells Syndrome and Bowen Armstrong Syndrome Andreea Liana Rachisan, MD; Simona Cainap, MD; Mariana Andreica, MD, and Nicolae Miu, MD Departement of Pediatrics, University of Medicine and Pharmacy "Iuliu Hatieganu", Cluj Napoca, Romania * Corresponding Author; Address: 2nd Clinic Of Pediatrics, Cluj Napoca, Romania 3-5 Crisan Street, Cluj Napoca 400177, Romania E-mail: andreea_rachisan@yahoo.com Received: Feb 26, 2010; Final Revision: May 17, 2010; Accepted: Jun 16, 2010 Code Number: pe11022 Abstract Background: Several
clinical entities combine ectodermal dysplasia (ED) and cleft lip and/or
palate (CL/P). These disorders have been recognized with a narrow phenotypic
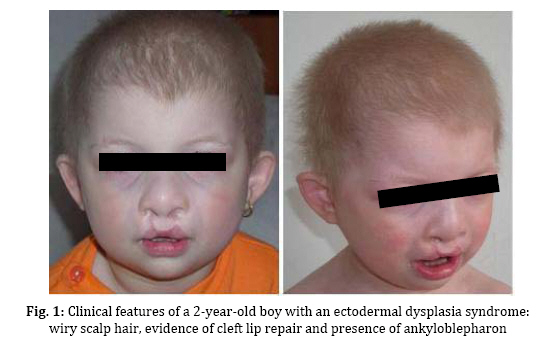
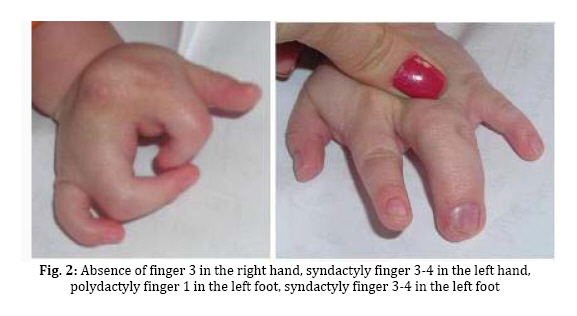
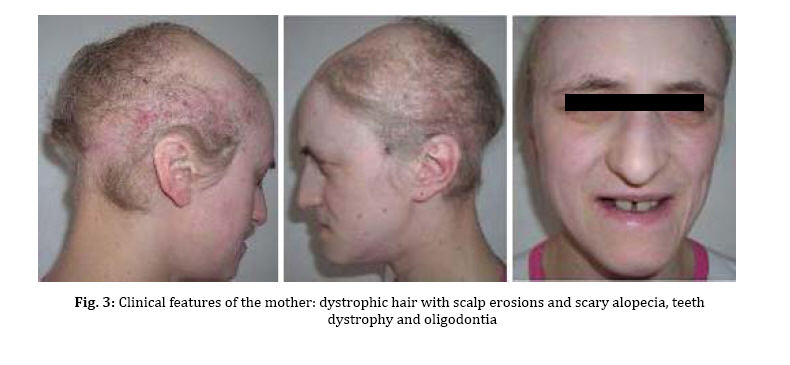
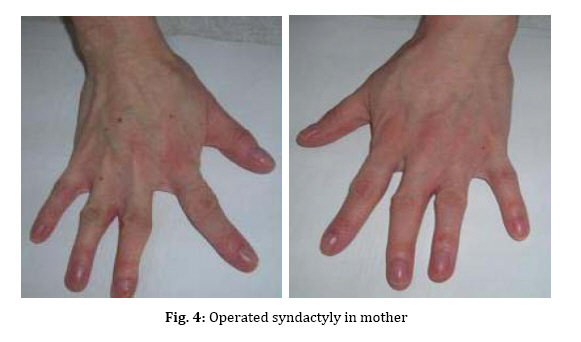
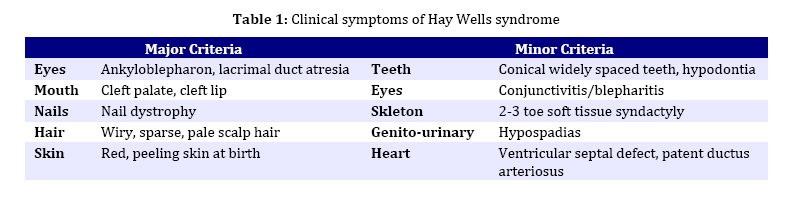
spectrum and very similar clinical features. Key Words: Ectodermal Dysplasia; Cleft Lip and/or Palate; Ankyloblepharon; Memtal Retardation Introduction The ectodermal dysplasia (ED) represents a complex of developmental abnormalities of skin, hair, nails, teeth and sweat glands. Several clinical disorders combine ED and cleft lip and/or palate (CL/P). The most commonly diagnosed are the AEC syndrome (ankyloblepharon, ectodermal abnormalities and CL/P), also known as Hay-Wells syndrome, the Bowen Armstrong syndrome (ED, CL/P and mental retardation) and the Rapp–Hodgkin syndrome (ED, CL/P and maxillary hypoplasia[1-3]. However, many of the reported clinical entities seem to be mutations of the same gene and often have overlapping and very similar clinical features[4]. Indeed over 30 syndromes have now been characterized with molecular genetics[5]. The Hay Wells syndrome was first described in 1976[6] and only about 50 cases have been reported todate. With an autosomal dominant inheritance and a variable clinical expressivity[7], the Hay Wells syndrome has a pathognomonic sign: the ankyloblepharon filiforme adnatum. This allows its differentiation from the other ED-CL/P syndromes. On the other hand, the Bowen Armstrong syndrome was described also in 1976[8] and until now 3 cases have been reported. In these patients the Hay Wells syndrome was excluded and the family referred in the literature as having the Bowen Armstrong syndrome[9]. Case Presentation The patient, a 2-year old boy, was the product of the second pregnancy of a young couple. The first child of this couple, a boy, deceased immediately after birth due to congenital malformations and affirmative with an ED-CL/P syndrome. The father is clinically healthy, without a positive history for congenital and genetic diseases. The mother has an ED-CL/P syndrome and is descendent of a non-consanguineous couple. She is the fifth child of the healthy couple and all the other sisters are clinically normal. The clinical examination of the boy revealed the typical symptoms of Hay Wells syndrome. He presented a characteristic face with a dystrophic hair and scalp dermatitis, the pathognomonic sign ankyloblepharon (adhesion of the ciliary edges of the eyelids), a median cleft palate repaired at the age of 12 months (Fig. 1).He also had dystrophic teeth and nails, bone malformations such as syndactyly of the fingers 3-4 in the left hand, the third finger was missing in the right hand, syndactyly of the fingers 3–4 in the left foot, polydactyly of the first finger in the left foot (Fig. 2). The urogenital malformation was represented by hypospadias . The chromosome analysis showed a normal 46, XY karyotype. Ophthalmological examination revealed obstruction of the lacrimal ducts and normal visual acuity. Dermatological examination revealed obstruction of the sweat gland ducts. Abdominal, pelvic and cardiac ultrasonography did not find any obvious malformations. The psychomotor development was normal for the age. The clinical examination of the mother showed an ectodermal dysplasia with wiry hair, scalp dermatitis, alopecia, operated CL/P, oligodontia (Fig. 3), operated syndactyly, dystrophic nails (Fig. 4), and moderate mental retardation. Discussion The ED- CL/P syndromes are a group of clinical entities that exhibit a narrow phenotypic spectrum with multiple clinical features[10]. Because of this, the classification of ED syndromes is difficult and several authors suggested that these disorders are phenotypic variants of a single genetic defect[11]. There are several clinical reports of overlapping features in ED syndromes, including families in which individual members resemble more than one particular entity[4]. Hay Wells syndrome is diagnosed by the presence of 3 major criteria, but many other miscellaneous features are usually found (Table 1). The differentiation between Hay Wells syndrome and Bowen Armstrong syndrome is often difficult, since they share very similar clinical features. Bowen Armstrong syndrome is characterized by dystrophic hair with recurrent scalp erosions and scarring alopecia, nail and teeth dystrophy and mild to severe mental retardation. The presence of ankyloblepharon is possible, but the absence of lacrimal duct anomalies makes the difference with Hay Wells syndrome. The variable mental retardation is present in Bowen Armstrong syndrome and is absent in all other ED-CL/P syndromes. Therefore, differentiation within the broad spectrum of related syndromes is made based on clinical symptoms. We sustain that the phenotypic overlap between our patient with Hay Wells syndrome and his mother with Bowen Armstrong syndrome may indicate that these two disorders are the same clinical entity, although an explanation for the presence or absence of ankyloblepharon is still lacking[12]. Regarding the mental retardation in Bowen Armstrong syndrome, the authors attributed it to the lack of intellectual stimulation. Nevertheless, the family known in the literature as having the Bowen Armstrong syndrome in the first and the third sister, the ankyloblepharon was described as well as variable mental retardation. Notably, our patient did not have a history of erosive scalp dermatitis, a characteristic feature of the Hay Wells syndrome, but his mother had an erosive dermatitis with a marked alopecia[13]. Given the diagnostic dilemma in our case, we reviewed the family history. The affected child had a brother who deceased immediately after birth due to an ED-CL/P syndrome. The father and his relatives were clinically normal; the parents and the four older sisters of the affected mother were also clinically normal. Although our patient has a number of clinical abnormalities that closely resemble many of the ectodermal dysplasia syndromes, it is difficult to assign him at one of the established entities. The clinical overlap between the two syndromes is supported by recent molecular studies showing that Hay Wells syndrome and Bowen Armstrong syndrome may result from almost identical mutations in the SAM domain of P63[14,15]. The presence of two affected individuals in the same family (mother and brother), showed that the allelic heterogeneity is responsible for the great clinical overlap between these two conditions. Conclusion Although ED syndromes are very rare conditions, they should be considered in our diagnosis in children with suggestive symptoms and positive family history. Our clinical findings indicate that it might not be justified to use the eponyms in referring to these ED syndromes. We hope this approach will contribute to a more accurate classification of ED syndromes, as well as improving the comprehension of this complex group of inherited disorders. References
Copyright 2011 - Iran Journal of Pediatrics The following images related to this document are available:Photo images[pe11022f4.jpg] [pe11022f3.jpg] [pe11022f1.jpg] [pe11022t1.jpg] [pe11022f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}